
Structural Biology Platform Facilitates Discovery of Glutathione S-Transferase Inhibitors, Offering Novel Pathways for Drug Development

Introduction:
WuXi AppTec scientists recently contributed to a study which led to the discovery and optimization of a novel series of GST inhibitors. The authors used affinity mediated DNA-encoded library selection against a privileged scaffold to identify a compound with potent inhibition against human glutathione S-transferase Mu 2. SAR studies, coupled with X-ray crystallography and structural analysis, revealed that the binding mode of this inhibitor is quite different from known GSH-like compounds. This novel finding opens up promising avenues for the development of targeted therapies for specific GST-related diseases and sets the stage for the creation of more potent GST inhibitors.
Background:
Glutathione S-transferase (GST) is an essential, multifunctional enzyme superfamily that includes a range of subtypes, playing a pivotal role in biochemical defense systems. Participating in glutathione binding reactions as a transferase, GST is widely dispersed across the cytoplasm of various cell types, acting as a critical component in the metabolism of numerous intrinsic and extrinsic toxins. Beyond its detoxifying role, GST also participates in multiple biological processes such as cell signaling, post-translational modification, and programmed cell senescence. Within mammalian cells, GST proteins encompass subtypes like α, ζ, θ, μ, π, σ, and ω, each presenting potential therapeutic targets across various disease areas.
Currently, a vast array of GST inhibitors, including Ethacrynic acid (EA) and its glutathione conjugates, have been developed. Early clinical research demonstrates that EA can significantly curtail tumor growth by targeting GSTP1. However, its diuretic nature and insufficient selectivity for GSTs hamper its clinical application in cancer treatment. TLK199, a glutathione tripeptide analog, has the potential to address myelodysplastic syndromes by activating JNK via GSTP1 inhibition. Nonetheless, the subpar target selectivity and chemical stability of these inhibitors restrict their use. As a result, there remains an ongoing need to develop new inhibitors with structural diversity and adaptability to various therapeutic targets.
Methods:
Protein crystallography, a crucial aspect of structural biology, has seen significant advancements, and novel techniques that have sparked a revolution within the field. Central to this method is protein crystallization, which forms orderly crystal structures. This approach utilizes the diffraction generated by X-rays within crystals, and applies mathematical physics to derive the structure from these diffraction patterns. Progress in protein crystallization technology, coupled with the refinement of X-ray crystallography, has facilitated the derivation of numerous three-dimensional protein structures. By determining the structures of these intricate macromolecules and observing their binding sites, we can deepen our understanding of the interactions between drugs and receptors. This knowledge aids in a more comprehensive understanding of protein inhibitors, conformational activators, cofactors, and enzyme catalytic or “active” sites at a molecular level, thus enabling the design of superior drugs and enhancing therapeutic efficacy.
Results:
Each subunit of GST contains two structurally distinct domains: the more conserved N-terminal domain, composed of beta-sheets and alpha-helices; and the C-terminal domain, composed of 4-7 alpha-helices, connected to the N-terminal domain by a short sequence of about 10 amino acids. Structural and sequence variations between different GSTs are mainly reflected in the C-terminal domain, which determines substrate specificity. Each GST isozyme has a positively charged substrate-binding pocket, divided into a GSH-binding site (G-site) and an adjacent hydrophobic co-substrate-binding site (H-site). The G-site is located at the N-terminus and is the specific binding site for glutathione (GSH). The hydroxyl group of the Tyr residue at this site forms a hydrogen bond with the thiol group of GSH, ionizing GSH to form a stable, highly active thiolate anion, which drives GST binding, peroxidase, and isomerase reactions. The H-site is located at the C-terminus and is the hydrophobic substrate-binding site, whose structural variations confer substrate selectivity.
In the course of studying structure-activity relationships, we obtained the cocrystal structure of inhibitor 16 and Schistosoma japonicum glutathione S-transferase (SjGST, PDB: 8GYD). To gain deeper insights into its binding behavior, we overlaid the cocrystal structure of 16/SjGST with the structure of SjSGT that contains IBZGSH (Figure 1a). IBZGSH, a GSH analog, has a binding mode akin to GSH, and its quinone skeleton forms a hydrogen bond with Ser107. Contrasted with IBZGSH, compound 16 occupies the G-site and H-site differently. We noted that the carboxyl group of compound 16 partakes in forming a hydrogen bond with water molecules in the pocket and Tyr7. The crucial o-sulfonamidophenol group resides in the pocket created by the C-terminal helix and loop, forms a salt bridge with Gln207, and stabilizes its molecular conformation through π-π interactions with Trp8/206. The 2-chlorothiazole group is lodged into the H-site. We measured the dihedral angle between the quinone skeleton and the thiazole ring to be 28.3°, and the distance between the S and N heteroatoms to be 2.8 Å. This conformation benefits the interaction of the thiazole group with the key residues Try7/111. The GST pocket exhibits a high electrostatic potential, indicating its preference for binding low pKa ligands. Although compound 16 and IBZGSH significantly overlap at the H-site, compound 16 does not adopt the same GSH tripeptide backbone as IBZGSH, suggesting new directions for the development of non-GSH analog GST inhibitors.
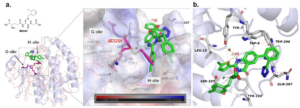
Figure 1. a) Overlap of the 16/SjGST cocrystal structure (PDB: 8GYD, with compound 16 depicted in green, SjGST in blue, and key residues shown in a gray sticks) with the IBZGSH/SjGST structure (PDB: 1M9B, where IBZGSH is shown in magenta and SjGST is represented by orange lines); b) Analysis of the binding model between compound 16 and SjGST (compound 16 is shown as green sticks, S atoms are yellow, N atoms are blue, and O atoms are red).
View the abstract of this publication by clicking HERE
Xin Wen et. al, 2023, Discovery, SAR Study of GST Inhibitors from a Novel Quinazolin-4(1H)-one Focused DNA-Encoded Library
Compound 16 has been identified as a potential GST inhibitor. It’s worth noting that the cocrystal structure of compound 16, when bound to SjGST, clearly shows that its binding mode considerably overlaps with IBZGSH at the H-site, thus significantly differing from known GSH-like compounds. This novel probe might operate via a mechanism distinct from GSH, potentially opening up new possibilities for designing more potent GST blockers. This finding is expected to contribute to the development of treatments targeting specific GST-related diseases.
These research outcomes further highlight the critical role of structural biology, specifically protein crystallography, in the discovery of drugs. Beyond its traditional applications in structure-based drug design, protein crystallography is now extensively employed to identify potential compounds, particularly during fragment-based screening. Without a doubt, protein crystallography will maintain a significant role in future drug development.
WuXi AppTec | Structural Biology Platform:
- High-throughput, automated screening platform;
- Extensive crystallization expertise, top-notch scientific team; more than 2000 commercial crystallization conditions;
- Comprehensive structural analysis service, starting from protein expression.
Related Content
Despite major advances in small molecule drug development, most disease-relevant proteins remain beyond the reach of conventional therapeutics. Molecular glues...
VIEW RESOURCEWith advancements in drug design, there is resurging interest in drugs that form covalent bonds with their targets. Covalent drugs...
VIEW RESOURCE