Physicochemical properties measurements
Drug leads should have sufficient target specificity and efficacy, however these properties alone are not enough. To be a druggable candidate for preclinical development, the compound should also have sufficient biocompatibility in terms of dissolution and absorption and favorable pharmacokinetics. Many pharmacologically active compounds fail even at this stage due poor bioavailability, unacceptable pharmacokinetics, or unexpected safety problems, some of which can be predicted based on the measured physicochemical properties. As a result, physicochemical parameters have been incorporated into drug discovery programs to rank the lead compounds and filter out unsuitable compounds. Properties such as pKa, solubility (KS, TS), and lipophilicity are among the most fundamental physicochemical traits of a drug candidate and their measurements are essential for both in silico and in vitro evaluation of drug-like properties.
WuXi AppTec’s methods for measuring common physicochemical properties
WuXi AppTec’s physicochemical property platform utilizes the following widely used methods for measuring pKa, solubility (KS, TS), and lipophilicity:
Physicochemical property | Measurable | Method |
Lipophilicity | logP | Scaled down shake flask method |
Potentiometric method | ||
logD | Shake flask method | |
pKa | pKa | Potentiometric method |
Spectrophotometric method | ||
Solubility | Kinetic solubility (K.S.) | Shake flask method |
Thermodynamic solubility (T.S.) | Shake flask method |
Solubility Assays – Kinetic Solubility and Thermodynamic solubility
The aqueous solubility of a drug substance is an important physicochemical parameter that has a significant role in various physical and biological processes including absorption, clearance and formulation. In rating the solubility of compounds for discovery project teams, compounds below 10 μg/mL are classified as poorly soluble, 10-60 μg/mL are moderately soluble and above 60 μg/mL are considered soluble. These solubility classification ranges can be suggested for medicinal chemists. And it is important to distinguish between “kinetic” and “thermodynamic” solubility.
Differences between kinetic and thermodynamic solubility.
Items | Kinetic solubility | Thermodynamic solubility |
Starting material | Initially fully dissolved in an organic solvent (e.g., DMSO) then added to the aqueous buffer | Solid crystalline material added into aqueous buffer |
Equilibrium | Not reached | Reached |
Effect of crystal form | Not related as it begins fully dissolved | The crystal form dictates solubility and dissolution rate |
Application | Early discovery stage to mimic the protocols for discovery assays that begin with DMSO stock solutions | Late discovery and early development where a large batch of API has been synthesized and its crystal form been characterized |
Discovery and development scientists have different needs when considering solubility. The goal of discovery scientists is to ensure they can obtain sufficient assay concentrations when they dilute a stock solution of a drug into a working buffer. In this case, amorphous and metastable crystal forms are not necessary to consider. During the development phase, the goal is to develop a viable drug formulation and perform the detailed technical studies required for regulatory approval, thus full characterization of the API is necessary. WuXi AppTec’s solubility platform provides both kinetic solubility and thermodynamic solubility assays. Stage of development will dictate which one is necessary for a given study.
pKa assay
Knowing and understanding the pKa of a compound is very important in drug discovery and development as it influences how a drug will behave in various bodily fluids. This makes it easier to foresee absorption and pharmacokinetic issues. WuXi AppTec’s pKa platform utilizes both potentiometric and spectrophotometric methods using a Sirius T3 instrument. The choice of whether to run a potentiometric or spectrophotometric assay depends on a number of factors including: the expected solubility of the compound, the amount of compound available, the expected pKa values and the UV absorbance characteristics of the drug.
Comparison of the two methods used to determine pKa.
Method | Description | TAT (working days) |
vPotentiometric method (pH-metric method) | • The standard working pH range is 3.0 to 11.0 | 5 |
Spectrophotometric method (UV absorbance method) | • The standard working pH range is 2.0 to 12.0. • 1 mg sample powder or 50µL of a 10 mM DMSO stock aolution is required | 5 |
As drug-like molecules are often poorly water soluble, a water-miscible co-solvent can be used to enhance solubility. By performing titrations at several different solvent/water ratios, the pKa in a purely aqueous environment can be extrapolated for water-insoluble compounds.
Lipophilicity – log P
log P
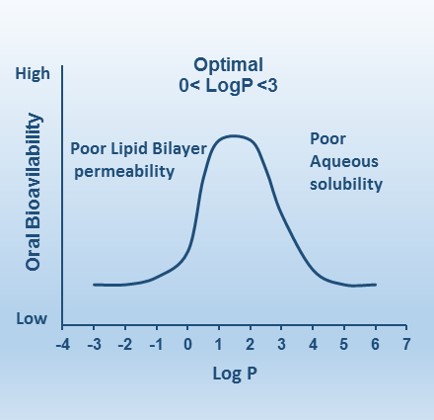
The lipophilicity of a compound dictates its partition coefficient (P); a measurement of the tendency to partition into a nonpolar lipid phase from an aqueous phase. It is an important determinant of most other drug properties and is a rapid and effective tool for an initial assessment of druggability. A general guide for optimal gastrointestinal absorption by passive diffusion following oral dosing is to have a moderate log P (between 0–3). In this range, a good balance of permeability and solubility exists. Compounds with a lower log P are more polar and while they typically have excellent solubility, they tend to have poorer lipid bilayer permeability. Thus these compounds will need to be small enough and preferentially nonionic or cationic to absorb paracellularly, utilize natural uptake transporters in the gut lumen or be administered parenterally. Compounds with a higher log P are more nonpolar and can thus suffer from dissolution and solubility issues as well as exhibiting a depot effect in lipids making it harder to clear the compound.
Lipophilicity – log D
Since the majority of drugs (approximately 80%) are ionizable, log P is not a sufficient predictor of a compound’s behavior across the physiological pH spectrum. Instead the log D is a more useful descriptor. Since the log D is pH-dependent, the pH of the aqueous phase should always be specified when reporting a log D value. Most commonly the log D at a pH of 7.4, the physiological pH of the vasculature, is used. Table 3 shows what various log D values at pH 7.4 suggest with respect to druggability.
log D at pH 7.4 | Typical Effect on Physiological Properties | Typical Effect on In Vivo Properties |
<1 | • High solubility | • Low Vd |
1 to 3 | • Moderate solubility | • Balanced Vd |
3 to 5 | • Low solubility | • High Vd • Oral absorption variable; may see lymphatic absorption |
> 5 | • Poor solubility | • High Vd and low CL due to depot effect • Dissolution limitations in GI |
General predictions on how the log D at pH 7.4 will affect the druggability of a compound.
Quality Control (QC)
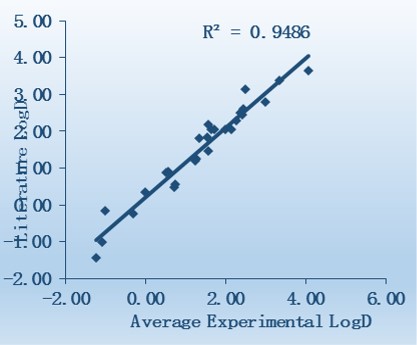
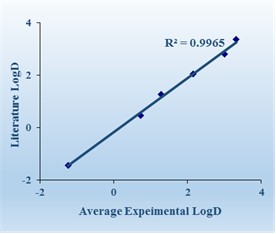
Nadolol, mexiletine, propranolol, quinidine, amitriptyline and chlorpromazine are used as assay standards for the log D assay.
Additional Validation Compounds
WuXi AppTec has screened 27 commercial drugs using our log D assay system. The results show the average experimental log D values correlate well with those reported in the literature.