
Targeting ER Mutations and Drug Resistance

Targeting ER Mutations and Drug Resistance: R&D Strategies and Comprehensive Preclinical Evaluation of Novel ER Degraders
Introduction
Estrogen receptor (ER)-positive is the most common subtype in breast cancer patients. Endocrine therapy, by inhibiting or blocking the estrogen-ER signaling pathway, serves as the cornerstone treatment for this type of cancer. However, in the face of various ESR1 mutant subtypes and increasingly prominent clinical drug resistance, developing next-generation inhibitors that cover different mutation types and effectively overcome resistance has become a key direction for enhancing the clinical value of ER-targeted therapies.
ESR1 Mutations: Challenges for Traditional Breast Cancer Therapies
In the field of breast cancer treatment, ER is a crucial therapeutic target. Approximately 70% of breast cancer patients are ER-positive, and the growth of their tumor cells is highly dependent on the estrogen-ER signaling pathway. For decades, endocrine therapy has been the cornerstone treatment for these patients by inhibiting estrogen synthesis or blocking ER function. However, drug resistance remains a core clinical challenge, among which mutations in the ESR1 gene, which encodes ERα, represent a key mechanism leading to acquired resistance [1].
ESR1 mutations primarily occur in the ligand-binding domain (e.g., Y537S, D538G), causing constant ER activation even in the absence of estrogen stimulation, thereby driving tumor growth [2]. Among advanced patients receiving endocrine therapies such as aromatase inhibitors, the detection rate of ESR1 mutations can reach 20%-40%, which is closely associated with a poor prognosis. Traditional selective estrogen receptor degraders (SERDs) like fulvestrant can degrade ER, but their efficacy against certain ESR1 mutants is limited. Furthermore, the requirement for intramuscular injection poses challenges to patient compliance.
ARV-471 PROTAC Brings New Hope to Breast Cancer Treatment
On May 1, 2026, Vepdegestrant (ARV-471), a PROTAC (Proteolysis Targeting Chimera) ER degrader co-developed by Pfizer and Arvinas, was approved by the US FDA for the treatment of ER+/HER2- advanced breast cancer. It became the world’s first approved PROTAC drug, marking a brand-new paradigm in ER-targeted therapy [3].
As an oral PROTAC degrader, ARV-471 features a molecular design that allows it to bind to both wild-type and multiple ESR1 mutant ER proteins with equal high efficiency. Preclinical studies have shown that, across various ER-positive breast cancer cell lines, it not only degrades wild-type ER but also demonstrates potent degradation capabilities against key clinical mutants, including Y537S, D538G, Y537C, Y537N, E380Q, L536P, and V422del, with degradation rates exceeding 90%. This “event-driven” degradation mechanism completely removes the oncogenic drivers at the protein level, rather than merely inhibiting their function, providing a fundamental solution to overcoming ESR1 mutation-mediated drug resistance.
Data from the pivotal Phase III VERITAC-2 study fully validated its breakthrough efficacy. Among 270 patients harboring ESR1 mutations, the median progression-free survival (PFS) in the ARV-471 group was 5.0 months, significantly superior to the 2.1 months in the fulvestrant control group, reducing the risk of disease progression or death by 43% (HR = 0.57). Additionally, the objective response rate (ORR) for ARV-471 was 18.6%, more than four times that of the control group (4.0%); the clinical benefit rate (CBR) reached 42.1%, also significantly higher than the control group’s 20.2%. Regarding safety, ARV-471 was well-tolerated. The most common adverse events included fatigue (27%) and elevated transaminases (14%), mostly Grades 1-2, and the discontinuation rate due to adverse events was low (2.9%) [4, 5].
The success of ARV-471 not only provides a highly effective new oral option for the difficult-to-treat patient population with ESR1 mutations, but also offers the first clinical validation of the immense potential of the revolutionary PROTAC technology, which may lead to a new direction in oncology treatment.
Diverse Drug-Resistant Tumor Models Empower Breast Cancer Drug R&D
Traditional endocrine therapies for ER+ breast cancer are limited by drug resistance, resulting in restricted therapeutic efficacy. The sources of resistance vary and can mainly be divided into two categories: ER-related mutations and growth factor / kinase pathway regulation.
ER-related mutations are primarily mutations in the ESR1 gene. Activating mutations in the ER ligand-binding domain (such as Y537S and D538G) lead to ligand-independent ER activity, enabling tumors to grow in the absence of estrogen, thereby inducing partial drug resistance and estrogen independence [6]. Clinically, these mutants can still be partially inhibited by endocrine therapies like fulvestrant, but they typically require more potent antagonists or next-generation selective estrogen receptor degraders (SERDs)/selective estrogen receptor modulators (SERMs). Another ER-related alteration is ER loss or reprogramming. Some fulvestrant-resistant models downregulate the ER signaling pathway and become estrogen-independent, exhibiting variable cross-resistance to tamoxifen.
The regulation of growth factor/kinase pathways is highly complex. The following mechanisms have been reported [7]:
- HER2 mutations: Acquired HER2 kinase-activating mutations in metastatic ER+ disease lead to estrogen independence and confer resistance to tamoxifen, fulvestrant, and CDK4/6 inhibitors. Combining ER therapy with the HER2 inhibitor neratinib can restore sensitivity.
- FGFR/FGF alterations: FGFR/FGF amplification or mutations drive resistance to fulvestrant and CDK4/6 inhibitors through ER “reprogramming” and MAPK activation. The use of FGFR, MEK, or SHP2 inhibitors can reverse this process.
- Broader pathway activation:The activation of PI3K/AKT/mTOR, RAS-MAPK, EGFR/HER2, IGF, and related pathways also mediates resistance to tamoxifen/aromatase inhibitors / SERDs and supports endocrine-independent tumor growth.
Case Study 1: Hit Discovery
WuXi Biology has established integrated drug discovery capabilities to accelerate the discovery of novel PROTACs. First, High-throughput screening (HTS) combined with powerful DNA-Encoded Library (DEL) technology is utilized to rapidly identify targeted ligands (warheads) that bind to target proteins and E3 ligases. Subsequently, Direct-to-Biology (D2B) technology is employed to conduct nanomole-scale high-throughput chemical reactions in microplates. This enables PROTACs to be batch-synthesized directly within the microplates and immediately used for biological testing. While significantly reducing the consumption of raw materials and reagents, it takes only 2–3 weeks to complete the design, synthesis, testing, and primary screening data analysis (Design-Make-Test-Analyze, DMTA cycle) for thousands of PROTACs, thereby significantly shortening the timelines for hit discovery and drug structural optimization (Figure 1).

Figure 1. D2B platform accelerates PROTAC discovery and optimization.
Case Study 2: In Vitro Protein Degradation Assay Platform
WuXi Biology has established an in vitro degradation assay platform specifically for the ER protein. By employing techniques such as Time-Resolved Fluorescence Resonance Energy Transfer (TR-FRET) and In-Cell Western Blot, this platform provides high-quality data support for the research and development of ER-targeted drugs (Figure 2). These assays deliver highly reproducible results with significant dose-responsiveness, offering the precision and sensitivity required for rigorous drug screening and optimization.

Figure 2. In Vitro protein degradation assay platform
Case Study 3: Efficacy of Standard Endocrine Therapies and Novel Degraders in MCF-7 ERα Y537S and D538G Models
WuXi Biology has established the MCF-7 ERα Y537S and MCF-7 ERα D538G mutant models. The established models have shown distinct phenotypes to ER+ therapies both in vitro and in vivo, providing platforms for ER mutant cancer treatment development. Utilizing these models, WuXi Biology systematically evaluated the in vivo anti-tumor activities of multiple ER+ breast cancer therapies (e.g., fulvestrant, tamoxifen, and ARV-471) (Figure 3).
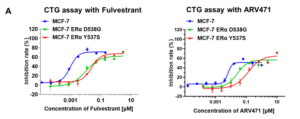
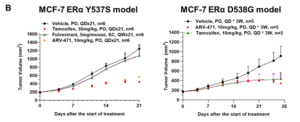
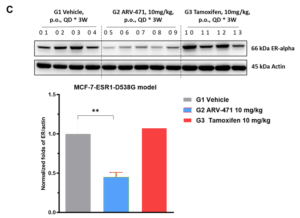
Figure 3. Efficacy of standard endocrine therapies and novel degraders in MCF-7 ERα Y537S and D538G models
Case Study 4: Fulvestrant- and Tamoxifen-Induced MCF-7 Resistant Models
WuXi Biology established a tamoxifen-resistant MCF-7 model through tamoxifen induction. Subsequently, through further induction with fulvestrant, we developed a fulvestrant/tamoxifen dual-resistant MCF-7 model (Figure 4), providing a critical preclinical platform to overcome resistance to ER-targeted therapies. This robust model facilitates the efficient screening of novel therapeutic agents and helps identify mechanisms underlying multi-drug resistance, thereby accelerating the development of next-generation ER-targeted treatments.

Figure 4. Fulvestrant- and tamoxifen-induced MCF-7 resistant models
Through engineering modifications and long-term drug induction, WuXi Biology has successfully established multiple ER-related mutant and drug-resistant models (Table 1). These models can be utilized to elucidate resistance mechanisms and evaluate the anti-tumor activity of next-generation inhibitors or combination regimens in resistant settings, thereby providing a critical preclinical platform for ER+ breast cancer research and for overcoming resistance to ER-targeted therapies.
Table 1. ER-related mutant and resistant models
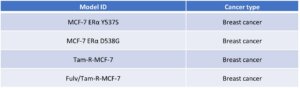
Based on a deep understanding of the biological mechanisms of the ER target and the technical requirements for drug discovery, WuXi Biology has established an end-to-end service platform for ER-targeted therapies. This platform covers in vitro biochemical assays, cell/animal models, and efficacy evaluation in drug-resistant models across different ER mutations and forms of endocrine resistance. WuXi Biology is committed to building a one-stop empowerment platform for ER-related breast cancer drug R&D, helping global partners accelerate the discovery and clinical translation of next-generation ER-targeted therapies.
References:
[1] Ferrari, P.; Schiavone, M.L.; Scatena, C.; Nicolini, A. Molecular Mechanisms and Therapeutic Strategies to Overcome Resistance to Endocrine Therapy and CDK4/6 Inhibitors in Advanced ER+/HER2− Breast Cancer. Int. J. Mol. Sci. 2025, 26(7), 3438.
[2] Brett, J.O., Spring, L.M., Bardia, A. et al. ESR1 mutation as an emerging clinical biomarker in metastatic hormone receptor-positive breast cancer. Breast Cancer Res 23(1), 85 (2021).
[3] FDA approves vepdegestrant for ER-positive, HER2-negative, ESR1-mutated advanced or metastatic breast cancer. News release. US Food and Drug Administration. May 1, 2026. Accessed May 1, 2026. https://tinyurl.com/zjm3x8rp
[4] Arvinas announces FDA acceptance of the new drug application for vepdegestrant for the treatment of ESR1m, ER+/HER2− advanced breast cancer. News release. Arvinas, Inc. August 8, 2025. Accessed May 1, 2026. https://tinyurl.com/3u6csmv9
[5] Hamilton EP, De Laurentiis M, Jhaveri KL, et al. Vepdegestrant, a PROTAC estrogen receptor (ER) degrader, vs fulvestrant in ER-positive/human epidermal growth factor receptor 2 (HER2)–negative advanced breast cancer: results of the global, randomized, phase 3 VERITAC-2 study. J Clin Oncol. 2025;43(suppl 17):LBA1000. doi:10.1200/JCO.2025.43.17_suppl.LBA1000
[6] Andrew Octavian, S., & Ying Pei, W. (2018). Organoids as Reliable Breast Cancer Study Models: An Update. International Journal of Oncology Research, 1:008.
[7] Bhushan, A.; Gonsalves, A.; Menon, J.U. Current State of Breast Cancer Diagnosis, Treatment, and Theranostics. Pharmaceutics 2021, 13(5), 723.
Related Content
OncoWuXi Express will continue to keep you informed of updates to our online pharmacology model database (OncoWuXi Database) and our...
VIEW RESOURCEBuilding on the momentum of AACR 2026, WuXi AppTec was honored to host an outstanding group of regional industry leaders...
VIEW RESOURCE