
Short-Term Mouse Model to Accelerate SLE Drug Discovery

Accelerating SLE Drug Discovery: A Short-Term BM12-Induced Mouse Model for Efficient Drug Screening
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease affecting about 5 million people worldwide, 90% of whom are women [1]. The disease can affect multiple organs, including the skin, joints, kidneys, heart, and nervous system, and can be life‑threatening in severe cases. Clinically, SLE is usually classified by the main affected organ, specifically including cutaneous, renal, neuropsychiatric, and other forms, and is further subdivided using immunologic markers such as anti-double stranded DNA (dsDNA) antibodies and hypocomplementemia to guide treatment [2]. In recent years, molecular feature–based subclassification has also been increasingly applied in clinical practice to help select targeted therapies for different pathogenic mechanisms.
The Pathogenesis of SLE
The onset of SLE occurs on a background of genetic susceptibility and epigenetic modifications caused by multigenic variants such as human leukocyte antigen (HLA), interferon regulatory factor 5 (IRF5), and signal transducer and activator of transcription 4 (STAT4), and is jointly triggered by various environmental factors, including estrogens, ultraviolet radiation, and Epstein-Barr virus (EBV) infection. Its central mechanism is the breakdown of immune tolerance, leading to abnormal activation of innate immune cells, particularly plasmacytoid dendritic cells (pDCs), which release large amounts of type I interferons (I-IFNs) and thereby drive dysregulation of T and B lymphocyte functions (Figure 1).
During this process, excessive B-cell proliferation and the spontaneous production of large amounts of pathogenic autoantibodies directed against nuclear components such as dsDNA, histones, and Sm antigens are key events. Follicular helper T cells (Tfh) undergo abnormal expansion and hyperactivation, persistently issuing “incorrect instructions” to B cells and driving them to produce copious pathogenic autoantibodies. Concurrently, T-cell subset imbalances-such as an increased Th17/regulatory T cell (Treg) ratio-and overexpression of B cell activating factor (BAFF) further exacerbate this abnormal autoimmune response.
Ultimately, large quantities of autoantibodies bind antigens to form immune complexes that deposit in multiple organs throughout the body; by activating the complement system and recruiting inflammatory cells, they provoke sustained inflammatory injury in tissues such as the kidney, skin, and joints. At the same time, impaired clearance of apoptotic cells and cellular debris by macrophages leads to ongoing release of self-antigens, creating a vicious cycle that worsens disease progression [3, 4].
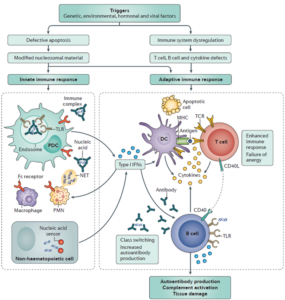
Figure 1. The pathogenesis of SLE[3]
Clinical Treatment Strategies and R&D Directions for SLE
The clinical management of systemic lupus erythematosus centers on a “treat-to-target” principle, aiming to minimize glucocorticoid exposure through individualized therapy. Hydroxychloroquine is the cornerstone medication for all patients and is commonly combined with glucocorticoids and traditional immunosuppressants such as mycophenolate mofetil [5, 6]. With the widespread use of biologics, belimumab and the domestically developed dual-target agent telitacicept have become important options for moderate and severe patients, effectively reducing steroid dependence and improving long-term outcomes.
The current R&D strategy of SLE drugs is rapidly evolving toward diversification and precision. Therapeutic strategies have moved beyond conventional broad immunosuppression. In biologics, focus has shifted from single B‑cell depletion (e.g., anti-CD20 antibodies) to novel formats such as bispecific antibodies, while strategies to directly eliminate long-lived plasma cells are being explored. At the same time, notable progress has been made with small molecules targeting core pathogenic pathways, such as oral inhibitors of type I interferon signaling, intracellular Toll-like receptors (TLRs), and Janus kinase/tyrosine kinase 2 (JAK/TYK2), which could provide new treatment options. Chimeric antigen receptor T-cell (CAR-T) therapy has shown potential for achieving “functional cures” in refractory patients and has become a research hotspot [7].

Table 1. Representative innovative drugs for SLE in clinical development worldwide [8]
Case study: A short‑course preclinical efficacy model for SLE
B cells play a critical role in the immunopathology of SLE, with functions extending beyond autoantibody production to antigen presentation, cytokine secretion, and regulation of T-cell responses [4]. The Immunology Center of WuXi Biology has established an in vitro B‑cell functional model for rapid screening of B-cell-targeting candidate drugs [9, 10].
B cells were isolated from peripheral blood mononuclear cells (PBMCs) and activated with anti‑immunoglobulin M (anti‑IgM) antibody plus interleukin‑4 (IL‑4), while either the BTK inhibitor ibrutinib or the BTK degrader bexobrutideg was added. Both agents markedly suppressed expression of the B‑cell surface activation marker CD69 (Fig. 2a) and release of the cytokine interleukin‑6 (IL‑6) (Fig. 2b). By inhibiting B‑cell function through blockade of the BTK signaling pathway, this study not only reveals a therapeutic mechanism but also provides a short‑course preclinical in vitro model for pharmacodynamic evaluation and potential biomarker screening in B‑cell‑targeted drug development.

Figure. 2. In vitro B‑cell activation and cytokine secretion
For SLE efficacy assessment, conventional animal models are categorized as spontaneous and induced:
- Spontaneous models include MRL/lpr, NZB/WF1, and Trex‑1 knockout mice;
- Induced models include TLR7 agonist and pristane models.
These in vivo models typically require about 10‑15 weeks to establish, which entails high R&D costs. To support early drug screening for SLE, WuXi Biology established a BM12‑induced SLE model that shortens the in vivo experimental period to 2 weeks. This model rapidly induces lupus‑like autoimmunity via adoptive T‑cell transfer. The core mechanism relies on minor differences in major histocompatibility complex class II (MHC II) between donor and recipient: CD4+ T cells from bm12 mice are transferred into genetically similar C57BL/6 recipients. Because the MHC II molecules differ by three amino acids, chronic graft‑versus‑host disease is induced, and its clinical and immunologic manifestations closely resemble human SLE.
This model reproduces multiple key pathological features similar to human SLE within just two weeks. Data show that model mice exhibit swollen spleen, increased kidney weights (Fig. 3a), and significantly elevated plasma anti‑dsDNA antibody (IgG/IgM) secretion (Fig. 3b). Expansion of B‑cell subsets, including germinal center B cells (GCB) and plasma cells, as well as Tfh cells, was also observed (Fig. 3c).
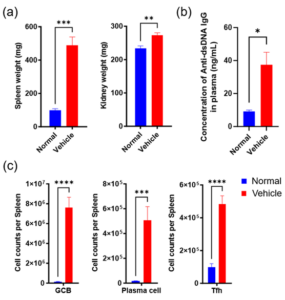
Figure 3. Pathological features of the BM12-induced SLE model.
The distribution and proportions of immune cells in plasma and spleen after induction closely match those seen in clinical SLE patients (Fig. 4), demonstrating that this model offers the advantages of a short timeline, SLE-like phenotypes, and high clinical relevance.
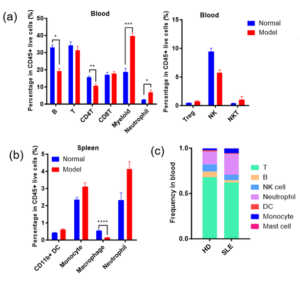
Figure 4. Changes of immune cells in the BM12-induced SLE model
Under treatment with the BTK inhibitor ibrutinib and the BTK degrader bexobrutideg, this SLE model exhibited marked pharmacodynamic responses at both cellular and protein levels. Both treatments significantly reduced production of the autoreactive anti‑dsDNA antibodies (Fig. 5a) and substantially decreased GCB numbers (Fig. 5b).

Figure 5. Regulation of autoantibodies and immune cell distribution by BTK inhibitor and degrader in the BM12‑induced SLE model
Ibrutinib and bexobrutideg significantly decreased multiple clinical biomarkers closely associated with SLE, such as interleukin‑6 (IL‑6), tumor necrosis factor‑alpha (TNF‑α), granulocyte‑macrophage colony‑stimulating factor (GM‑CSF), and interferon‑alpha (IFN‑α) (Fig. 6). Changes in these factors not only directly reflect the drugs’ effective suppression of systemic inflammation and immune dysregulation in the model, but given their high concordance with human SLE clinical biomarkers—underscore the model’s translational value for drug R&D and pharmacodynamic biomarker screening.
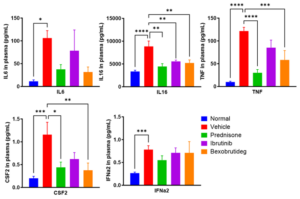
Figure 6. BTK inhibitors and degraders reduce levels of clinical biomarkers in the BM12-induced SLE model.
Summary
The BM12-induced SLE model reproduces disease-relevant changes at the cellular and protein levels. It elevates SLE-associated immune cell populations (GCB and Tfh) and anti-dsDNA antibody titers, and replicates increases in clinical biomarkers. The BM12-induced SLE model shortens in vivo experimental timelines from the conventional 10-15 weeks to about 2 weeks, significantly improving the efficiency of early drug screening. It should be noted that given the speed with which disease develops and its magnitude, the nephritis is mild in this model.
WuXi Biology Immunology Center supports the R&D of autoimmune drugs
Focusing on SLE pathogenic mechanisms, WuXi Biology has developed multiple in vitro assays targeting relevant pathways and cell types, including TLR7/8/9, cGAS-STING, inflammasomes, neutrophils, B cells, and T cells using cell lines and primary cells. WuXi Biology has also established various spontaneous and induced SLE animal models to support early and preclinical efficacy studies. Beyond SLE, the group has mature mouse, rat, and non-human primate (NHP) models for pharmacology and biomarker research in diseases such as atopic dermatitis (AD), psoriasis, rheumatoid arthritis (RA), inflammatory bowel disease (IBD), and Sjögren’s syndrome.
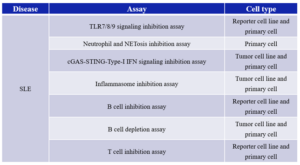
Table 2. Assays for SLE established by WuXi Biology Immunology Center
References
- Somers, Emily C et al. “Population-based incidence and prevalence of systemic lupus erythematosus: the Michigan Lupus Epidemiology and Surveillance program.” Arthritis & rheumatology (Hoboken, N.J.) vol. 66,2 (2014): 369-78. doi:10.1002/art.38238
- Franklyn, Kate et al. “Definition and initial validation of a Lupus Low Disease Activity State (LLDAS).” Annals of the rheumatic diseases vol. 75,9 (2016): 1615-21. doi:10.1136/annrheumdis-2015-207726
- Kaul, Arvind et al. “Systemic lupus erythematosus.” Nature Reviews. Disease primers vol. 2 16039. 16 Jun. 2016, doi:10.1038/nrdp.2016.39
- Dai, Xiaofeng et al. “Systemic lupus erythematosus: updated insights on the pathogenesis, diagnosis, prevention and therapeutics.” Signal transduction and targeted therapy vol. 10,1 102. 17 Mar. 2025, doi:10.1038/s41392-025-02168-0
- Fanouriakis, Antonis et al. “EULAR recommendations for the management of systemic lupus erythematosus with kidney involvement: 2025 update.” Annals of the rheumatic diseases vol. 85,1 (2026): 75-90. doi:10.1016/j.ard.2025.09.007
- Atacicept for Injection: Package Inset Reference
- Wang, Qian et al. “In Vivo CD19 CAR T-Cell Therapy for Refractory Systemic Lupus Erythematosus.” The New England journal of medicine vol. 393,15 (2025): 1542-1544. doi:10.1056/NEJMc2509522
- Dai, Xiaofeng et al. “Systemic lupus erythematosus: updated insights on the pathogenesis, diagnosis, prevention and therapeutics.” Signal transduction and targeted therapy vol. 10,1 102. 17 Mar. 2025, doi:10.1038/s41392-025-02168-0
- Morris SC, Cohen PL, Eisenberg RA. Experimental induction of systemic lupus erythematosus by recognition of foreign Ia. Clin Immunol Immunopathol. 1990 Nov;57(2):263-73. doi: 10.1016/0090-1229(90)90040-w.
- Klarquist J, Janssen EM. The bm12 Inducible Model of Systemic Lupus Erythematosus (SLE) in C57BL/6 Mice. J Vis Exp. 2015 Nov 1;(105):e53319. doi: 10.3791/53319.
Related Content
Strategies for Novel Autoimmune Drug Development: Preclinical Efficacy and Case Studies of JAK-STAT Targeted Therapies JAK Family Molecules: Central Hubs...
VIEW RESOURCEInflammatory bowel disease (IBD), which includes ulcerative colitis and Crohn’s disease, is a chronic, immune-mediated disorder characterized by persistent inflammation...
VIEW RESOURCE