
Advancements in Membranous Nephropathy
From Pathogenesis to Therapeutic Innovations
Introduction:
Membranous nephropathy (MN) has received particular attention among autoimmune kidney disease due to its distinct pathophysiology and clinical manifestations. Unlike other autoimmune nephropathies, MN is primarily driven by pathogenic autoantibodies that activate immune cells, leading to oxidative stress and inflammatory responses that compromise the glomerular filtration barrier [1, 2].
This article focuses on MN, providing a detailed exploration of its pathogenesis, recent advancements in drug development, and an introduction to the classic preclinical animal model widely used in MN research—the Passive Heymann Nephritis (PHN) model, along with a case study illustrating its application.
Types and Pathological Features of MN
MN is characterized by the deposition of immune complexes along the glomerular basement membrane (GBM) driven by autoantibodies, and typically manifests as nephrotic syndrome. MN can be classified into the following two categories:
- Primary Membranous Nephropathy (PMN): PMN is not associated with any underlying identifiable systemic diseases or drug exposures and presents as a kidney-limited condition, accounting for approximately 70% of cases. PMN is typically caused by autoantibodies targeting podocyte antigens, primarily the phospholipase A2 receptor (PLA2R) and, less commonly, thrombospondin type-1 domain-containing 7A (THSD7A).
- Secondary Membranous Nephropathy (SMN): SMN is associated with diseases or exposures that are linked to MN and may be causative factors (such as systemic diseases, infections, medications, or other underlying conditions), accounting for about 30% of cases. SMN is usually caused by the accumulation of exogenous antigens or epitopes in the subepithelial region of the basement membrane, which then become targets for circulating antibodies.
Progresses in MN Drug Development
In pathological processes, antigen-presenting cells (APCs) recognize and present podocyte-specific antigens, such as PLA2R, triggering T-cell and B-cell activation. Activated B cells differentiate into plasma cells, which produce autoantibodies that form immune complexes and deposit within the GBM. This deposition initiates complement activation, further damaging the filtration barrier and podocytes. As a result, glomerular permeability increases, leading to excessive proteinuria and the onset of MN. Persistent autoantibody production and complement activation contribute to GBM thickening, podocyte foot process effacement, and glomerulosclerosis, ultimately driving chronic kidney injury and disease progression (Figure 1) [5].
Rituximab, used off-label, is the only available targeted therapy for MN. It works by binding to the CD20 antigen on B cells, leading to B-cell depletion and reduced immune complex formation. Ongoing MN drug development efforts primarily focus on CD20, CD38, and BTK as therapeutic targets. Promising investigational therapies include CD20 monoclonal antibodies such as obinutuzumab and MIL62 (both in Phase III clinical trials) and the BTK inhibitor zanubrutinib (in Phase II/III clinical trials) [3,4].
A promising investigational drug is iptacopan (LNP023), which targets the complement system by inhibiting the alternative complement pathway. As a selective complement factor B inhibitor, iptacopan reduces excessive complement activation, thereby mitigating inflammation and tissue damage. This mechanism makes it a potential therapy for both MN and IgA nephropathy (IgAN). In a Phase II clinical trial for MN, iptacopan significantly reduced proteinuria and exhibited good toleration. A Phase III trial is underway to further assess its efficacy and safety in a broader patient population [6,7].
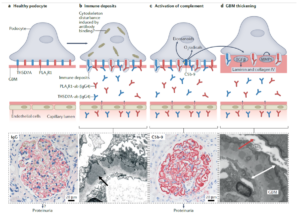
Figure 1. Pathogenesis of MN [6]
Preclinical Animal Models of MN
Animal models are essential for Autoimmune Kidney Disease (AIKD) research and drug development, serving as a critical bridge between basic research and clinical applications. By using various animal models, researchers can replicate the pathological processes of human autoimmune nephropathies, gaining deeper insights into disease mechanisms and progression. These models provide valuable platforms for identifying potential therapeutic targets, assessing the efficacy of novel treatments, and evaluating drug safety.
PHN is a well-established experimental model of nephropathy in which immune complexes deposit on the surface of renal tubular epithelial cells, triggering complement activation—particularly the formation of C5b-9, a membrane attack complex. This immune-mediated process leads to podocyte injury and proteinuria, closely mimicking the pathogenesis of human idiopathic membranous nephropathy (IMN). Due to its high translational relevance, the PHN model is widely used to investigate disease mechanisms and evaluate therapeutic interventions for membranous nephropathy.
The Renal Disease team at WuXi Biology has successfully developed a PHN rat model, selecting LNP023 as a representative candidate for testing.
Establishment of the PHN Model and Drug Efficacy Evaluation
To assess drug efficacy, rats were randomly divided into three groups: a healthy control group (untreated rats), a modeling group (rats were injected with anti-Fx1A serum to induce PHN) and a drug testing group (rats were injected with anti-Fx1A serum followed by LNP023 administration). Following anti-Fx1A serum injection, the urinary protein-to-creatinine ratio (uPCR) was examined, and it was significantly elevated in the modeling group which is similar to the clinical progression of MN. Notably, LNP023 treatment effectively and significantly reduced uPCR levels in PHN rats, demonstrating its potential therapeutic effect on MN.
Consistent with existing literature, serum creatinine and blood urea nitrogen (BUN) levels remained unchanged during the first two months post-PHN induction (Figure 2). However, in published chronic PHN models (2–18 months), a progressive increase in creatinine and BUN was observed over time [8–10].

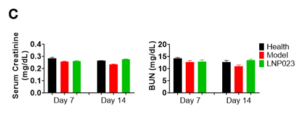
Figure 2. Characteristics of the PHN Model and Biochemical Parameter Alterations
Pathological Features in the PHN Model
In MN, histopathological changes—such as the hyperplasia of glomerular endothelium, GBM thickening, tubulointerstitial fibrosis and inflammatory cell infiltration occur. These histological changes are critical indicators for the successful establishment of the anti-Fx1A membranous nephropathy model.
Consistent with clinical features of MN, H&E staining (Figure 3A) revealed a significant increase in inflammatory cell infiltration (marked by the yellow arrow) in the model group, along with notable tubular injury (marked by the green arrow) and GBM thickening (marked by the red arrow).
In contrast, treatment with LNP023 significantly reduced local inflammatory cell infiltration, alleviated tubular damage, and improved GBM thickening and tubulointerstitial fibrosis compared to the model group (Figure 3B and 3C).

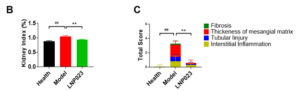
Figure 3. H&E Staining Illustrating the Pathological Features of the PHN Model
Complement System Activation in the PHN Model
C3, a key component of the complement system, plays a central role in immune-mediated kidney injury. Increased C3 deposition in renal tissues is an indicator of complement activation, which correlates with enhanced immune cell infiltration and tissue damage.
Immunofluorescence analysis (Figure 4A) revealed minimal C3 deposition in the glomeruli (marked by the arrow) of the healthy control group, significant C3 accumulation in the model group, and a marked reduction in C3 deposition following LNP023 treatment.
Statistical analysis confirmed that C3 deposition was significantly elevated in the model group but was markedly reduced with LNP023 treatment. Consistently, ELISA quantification of renal C3 levels demonstrated a significant increase in the model group compared to the healthy controls, while LNP023 administration effectively reduced C3 accumulation in the kidney (Figure 4B and 4C).

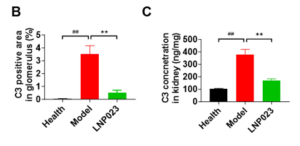
Figure 4. C3 Deposition Levels in the Kidney in the PHN Model
Immune Response in the PHN Model
In clinical practice, IgG intensity serves as a key marker of disease active status, as IgG deposition in the kidneys reflects complement activation and the involvement of the immune system. Strong IgG staining typically correlates with a stronger immune response, with severe kidney damage and higher disease activity.
Immunofluorescence analysis (Figure 5A) showed a significant increase in IgG deposition in the glomeruli of the model group compared to the healthy controls. However, the LNP023 treatment significantly reduced IgG accumulation as confirmed by statistical analysis (Figure 5B). These findings highlight the utility of the anti-Fx1A serum induced nephropathy model in preclinical research and underscore the protective effects of LNP023 in anti-Fx1A membranous nephropathy.
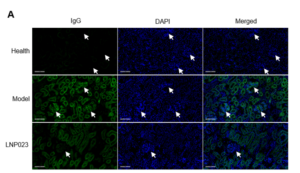
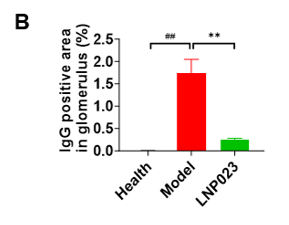
Figure 5. IgG Deposition in the Kidney in the PHN Model
A Final Word
The development of novel therapies for MN is paving the way for safer and more effective treatment options. Innovative biologic therapies and precision immunomodulation have significantly improved disease management, offering new therapeutic possibilities.
WuXi Biology has successfully established multiple AIKD animal models, including those for MN, to facilitate in-depth mechanistic research and drug discovery. Through continuous innovation and cross-disciplinary collaboration, we remain committed to accelerating breakthroughs in AIKD treatment and bringing new hope to patients worldwide.
References:
- Suárez-Fueyo, A., Bradley, S. J., Klatzmann, D., & Tsokos, G. C. (2017). T cells and autoimmune kidney disease. Nature reviews. Nephrology, 13(6), 329–343. https://doi.org/10.1038/nrneph.2017.34
- www.mayoclinic.org/diseases-conditions/nephrotic-syndrome/symptoms-causes/syc-20375608
- Kardaun, S. H., & Kuiper, H. (2023). Autoinflammatory keratinization diseases—The concept, pathophysiology, and therapy. Clinical Reviews in Allergy & Immunology, 64(1), 1–15. https://doi.org/10.1007/s12016-023-08971-3
- Hua, M. R., Zhao, Y. L., Yang, J. Z., Zou, L., Zhao, Y. Y., & Li, X. (2023). Membranous nephropathy: Mechanistic insights and therapeutic perspectives. International immunopharmacology, 120, 110317. https://doi.org/10.1016/j.intimp.2023.110317
- Hoxha, E., Reinhard, L. & Stahl, R.A.K. Membranous nephropathy: new pathogenic mechanisms and their clinical implications. Nat Rev Nephrol 18, 466–478 (2022). https://doi.org/10.1038/s41581-022-00564-1
- ‘ClinicalTrials.gov’ – APOLLO Trial
- ‘Novartis.com’
- Salant DJ, Belok S, Madaio MP, Couser WG. A new role for complement in experimental membranous nephropathy in rats. J Clin Invest. 1980 Dec 1; 66(6):1339–50.
- Iversen BM, Ofstad J. Loss of renal blood flow autoregulation in chronic glomerulonephritic rats. Am J Physiol-Ren Physiol [Internet]. 1988 Feb 1
- Michael J. CoyneID, A. Eric Schultze, Donald J. McCrann, III, Rachel E. Murphy, Julie Cross, Marilyn Strong-Townsend, Corie Drake, Rebekah Mack. Evaluation of renal injury and function biomarkers, including symmetric dimethylarginine (SDMA), in the rat passive Heymann nephritis (PHN) model. PLOS ONE. 2022; 17(5): e0269085. https:// doi.org/10.1371/journal.pone.0269085
Related Content
Strategies for Novel Autoimmune Drug Development: Preclinical Efficacy and Case Studies of JAK-STAT Targeted Therapies JAK Family Molecules: Central Hubs...
VIEW RESOURCEAccelerating SLE Drug Discovery: A Short-Term BM12-Induced Mouse Model for Efficient Drug Screening Systemic lupus erythematosus (SLE) is a chronic...
VIEW RESOURCE