
Mechanism of a Highly Selective N2 Alkylation of Indazole
QM Magic Class | Chapter 50
Liting Dong, Qiuyue Wang, Yongsheng Chen, John S. Wai
< Magical Power of Quantum Mechanics
Pfizer chemists reported in 2022 a highly selective N2 alkylation of indazoles with primary, secondary, and tertiary alkyl 2,2,2-trichloroacetimidate in Synthesis[1]. Diverse 1H-indazoles and azaindazoles are selectively alkylated at N2 and no N1 isomer was observed. The reaction mechanism proposed by the authors, as shown in Figure 1, involves the alkyl 2,2,2-trichloroacetimidate undergoing protonation on the imide under acidic conditions. This activation facilitates nucleophilic displacement by the N2-indazole nitrogen, leading to the desired product after re-aromatization. We conducted quantum mechanical (QM) analyses to understand the high selectivity for N2 alkylation.
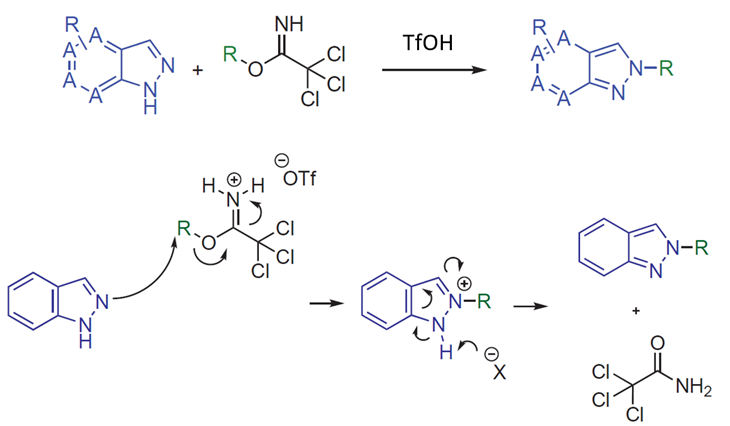
Figure 1. Proposed reaction mechanism for primary/secondary and tertiary alkyl 2,2,2-trichloroacetimidates (R is primary, secondary, and tertiary alkyl groups) [2]
QM analysis of the reaction mechanism

Initially, we selected two relatively simple structures in the article, indazole and methyl richloroacetimidate as models. Using Spartan 20’(DFT B97X-D/6-31G*), we calculated the reaction energy profiles for N1 and 2 alkylation. We estimated that the activation energy for N1 alkylation is 12.76 kcal/mol (Figure 2), and for N2 alkylation, it is at 13.87 kcal/mol (Figure 3). However, the lower activation energy for N1 alkylation by 1.11 kcal/mol contradicts the experimental observation of selective N2 alkylation. Th is discrepancy led us to explore the tautomeric forms of the indazole substrate, namely 1-H indazole and 2-H indazole, to understand their influence on the reaction selectivity.
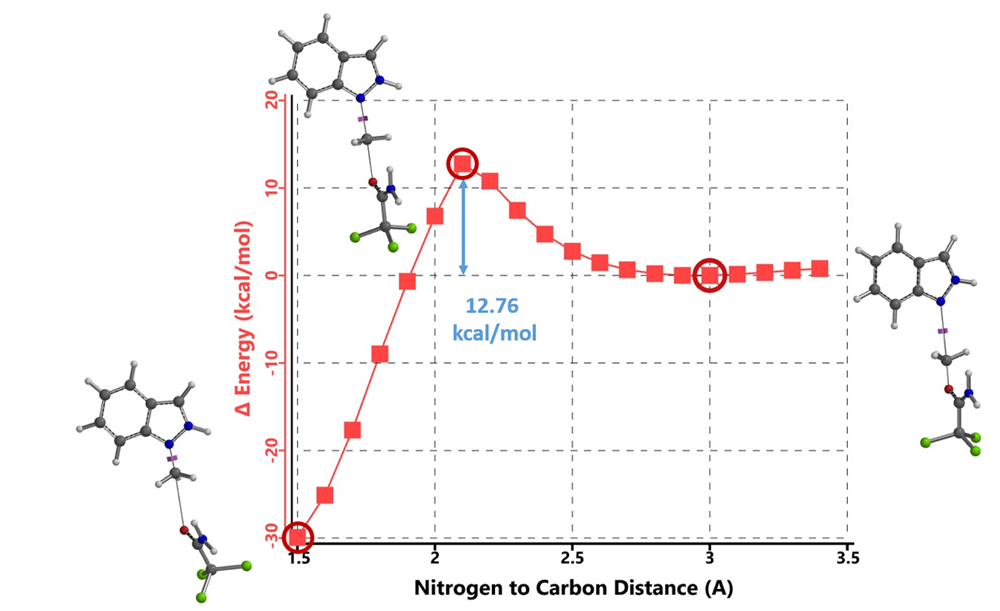
Figure 2. Reaction Energy Profile of N1 alkylation of Indazole
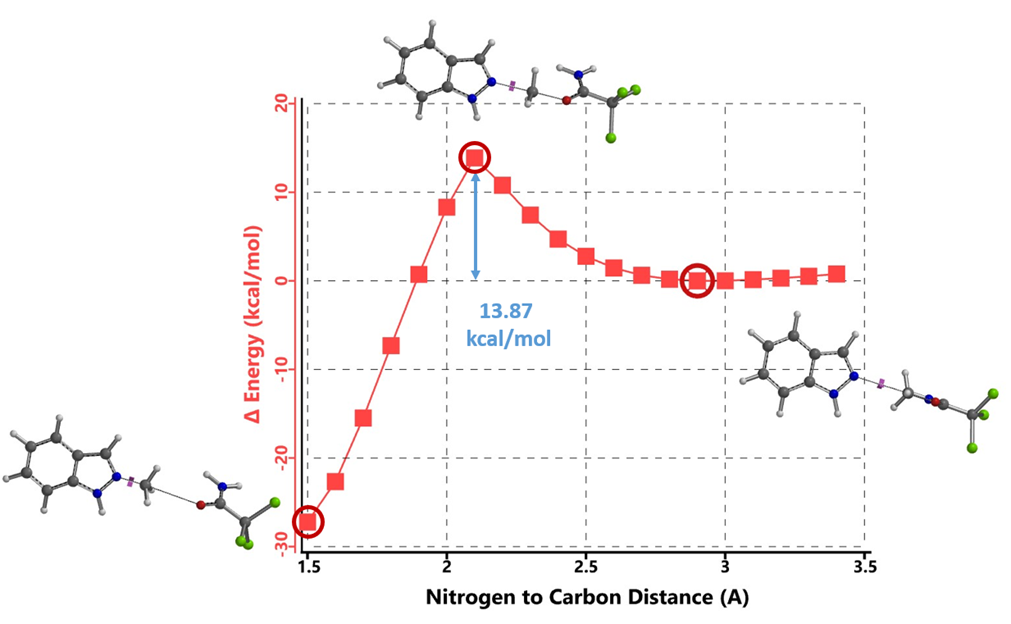
Figure 3. Reaction Energy Profile of N2 alkylation of Indazole
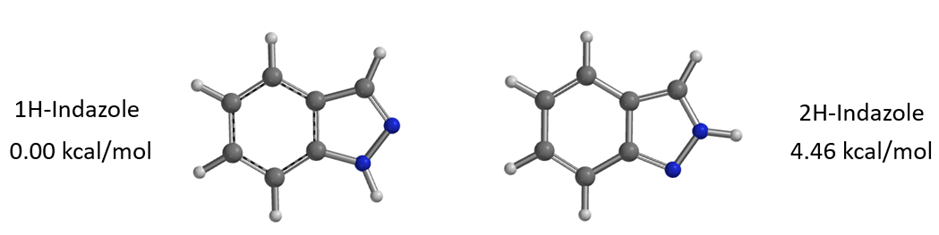
Figure 4. Relative energy of 1H- vs 2H-indazole tautomer
Calculations reveal that 1-H indazole is energetically more stable than its 2-H indazole tautomer by 4.46 kcal/mol (Figure 4). This energy difference must be included into the analysis of the two competing paths (Figure 5). For N1 alkylation, the substrate must convert from the lower-energy 1-H tautomer to the higher-energy 2-H form, which incurs an additional energy cost. Consequently, the total reaction energy barrier for N1 alkylation is 17.22 kcal/mol, which is 3.35 kcal/mol higher than that for N2 alkylation. This accounts for the high N2 selectivity observed (Figure 7)[3].
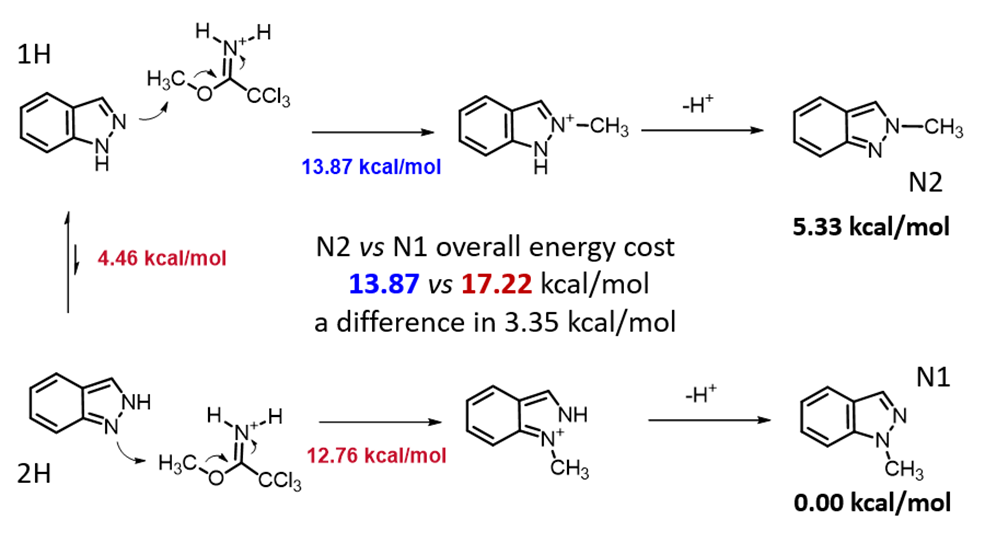
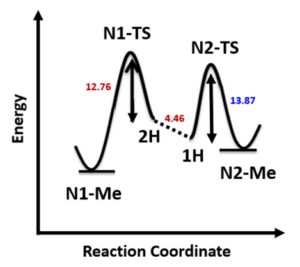
Figure 5. Comparison of energy changes in N1 vs N2 alkylation of Indazole
Transition state energy comparison
Based on the geometries at the highest energy points from the two reaction energy profiles (Figures 2 & 3), we calculated the relative energies of the transition states (TS) for further comparison. Each calculated TS exhibited only one imaginary frequency, corresponding to the vibration of the bonds being formed and broken (Figure 6)[4]. The energy difference between the N1 and N2 transition states is 3.42 kcal/mol, favoring N2. This is consistent with the earlier estimated energy difference that accounts for the observed N2 selectivity.
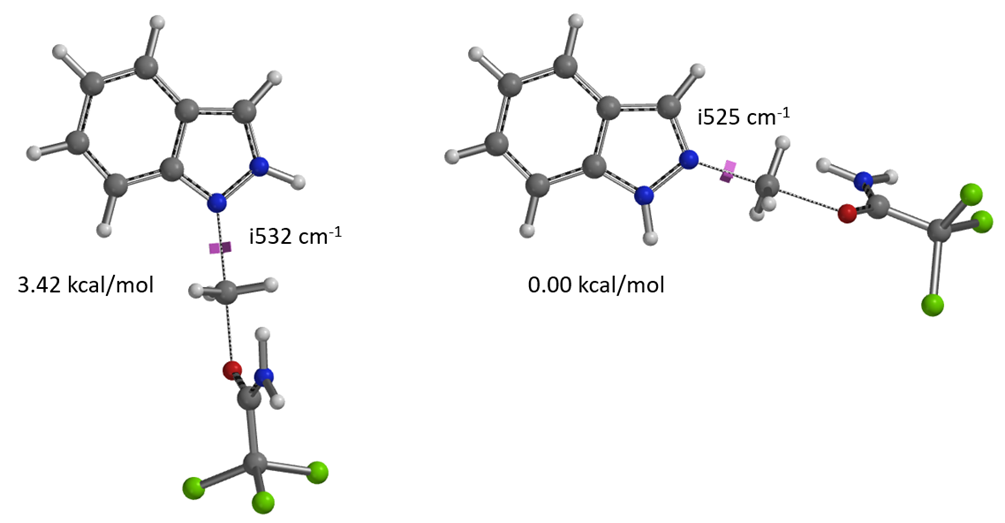
Figure 6. Relative energy comparison of indazole N1 and N2 alkylation transition states
The difference translates to an N2 vs N1 product ratio of 322:1 for a reaction at 25 °C, aligning with experimental results. With the energy difference between the TS greater than 3 kcal/mol, there is no practical difference if the reaction was run under reflux (dioxane b.p. 101 °C). The calculated ratio of N2 vs N1 products is still as high as 100 to 1 (Figure 7). This demonstrates an intrinsic high selectivity for N2-alkylation.
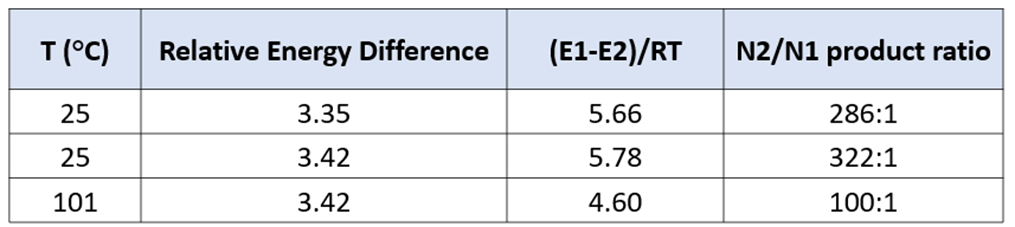
Figure 7. Product ratio calculated for the reaction at 25 and 101 °C
Substituted indazoles
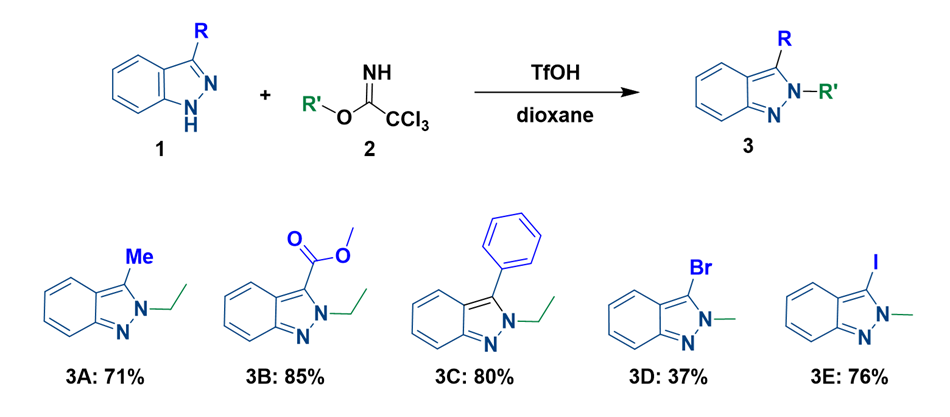
The paper reported that alkylation of indazoles with large C3 substituents (compounds 3A to 3C) remains highly N2 selective. We were interested in alkylation of 3-bromo and 3-iodo-indazole (compounds 3D & 3E) and found them to exhibit high N2 selectivity as well.
Table 1 presents the calculated relative energies of the 1-H and 2-H tautomers with various C3 substitutions as shown in column 3. The 2-H tautomers are consistently higher in energy, approximately 4 kcal/mol compared to the corresponding 1-H tautomers. This energy difference is even more pronounced for the 3-bromo and 3-iodo indazoles, reaching around 6 kcal/mol.
Activation energy calculated (by adding the energy difference between the tautomers and energy barrier from reaction energy profile calculations) for N1 alkylation is usually higher than that for N2 alkylation (column 6). However, there are outliers (highlighted in red) that are not consistent with observations. We attributed this to the less precise estimates of relative energies between tautomers[5]. In contrast, calculated transition state energy differences consistently correlate with experimental observations, revealing that N2 alkylation is generally favored by >3 kcal/mol, (column 7). For 3-tert-butylindazole, the difference is smaller, presumably due to steric effect.
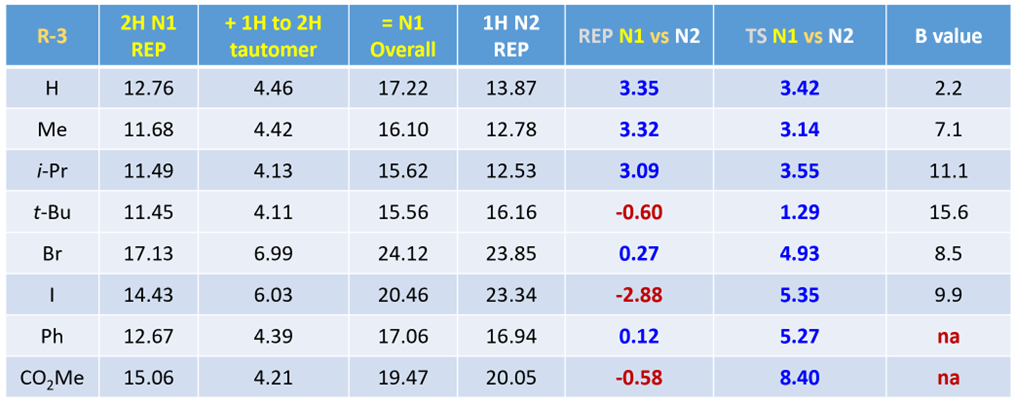
Table 1. Calculated data for alkylation of indazole with different C3 substituents
For indazoles with simple C3 alkyl groups, the relative energy difference between the TS for N1 and N2 alkylation correlates with the B values – a parameter established to quantify steric effects[6]. As B value increases, which indicates greater steric hindrance from the substituent, the energy difference becomes smaller. However, for 3-bromo and 3-iodoindazole, their corresponding B values would be suggestive of smaller energy differences between the N2 and N1 TS than the calculated ones. This can be attributed to a non-covalent interaction between the bromo or iodo group and NH of the protonated imidate, which stabilizes the N2 alkylation TS, increasing the energy difference between the two TS. This could be illustrated with the electrostatic potential (ESP) surfaces of the reacting partners (left side of Figure 9), with notable electrostatic interaction between the iodine atom of 3-iodoindazole and the imine hydrogen of the protonated imidate, akin to a halogen bond (right side of Figure 9)[7].
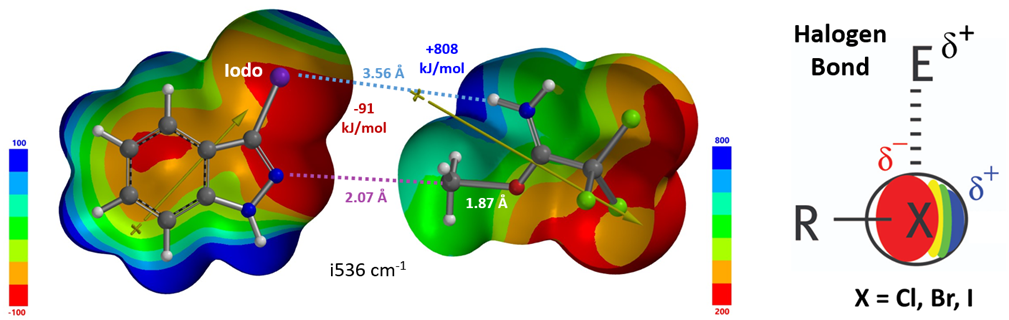
Figure 9. TS of 3-iodoindazole N 2 alkylation TS: ESP surfaces & dipole moments of indazole and imidate ester[8]
Although B values for phenyl and methoxycarbonyl groups were not reported[6], our calculations revealed the presence of non-covalent interactions (NCI) in the transition states (TS) of N2 alkylation of both the phenyl and ester substrates. Specifically:
1. Phenyl indazole TS: The electron density surface (EDS) of the TS (Figure 10, left) indicates an NCI between the C3 carbon of the phenyl group and the iminium H[9]. This interaction lowers the energy of the TS, facilitating N2 alkylation.
2. Methyl indazole-3-carboxylate TS: The EDS reveals a strong hydrogen bond between the carbonyl oxygen of and iminium H in the N2 TS (Figure 10, right), resulting in the highest energy difference between N1 and N2 TS of 8.40 kcal/mol in this study.
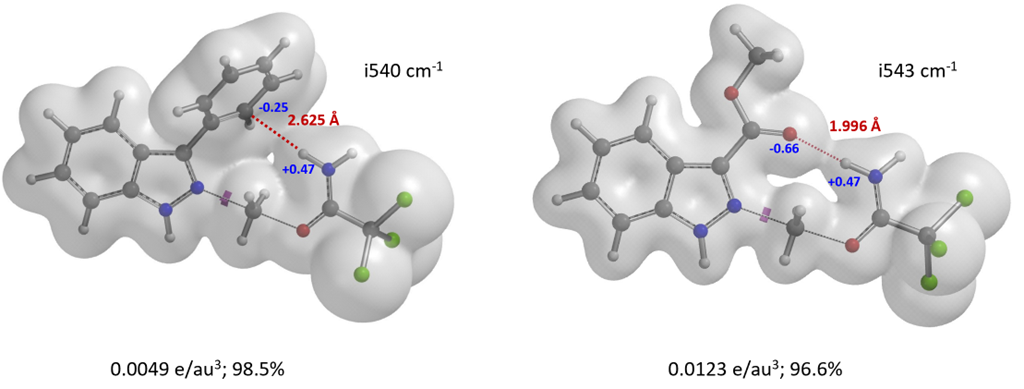
Figure 10. Electron density surface of the N2 alkylation TS: 3-phenylindazole & methyl indazole-3-carboxylate
Azaindazoles
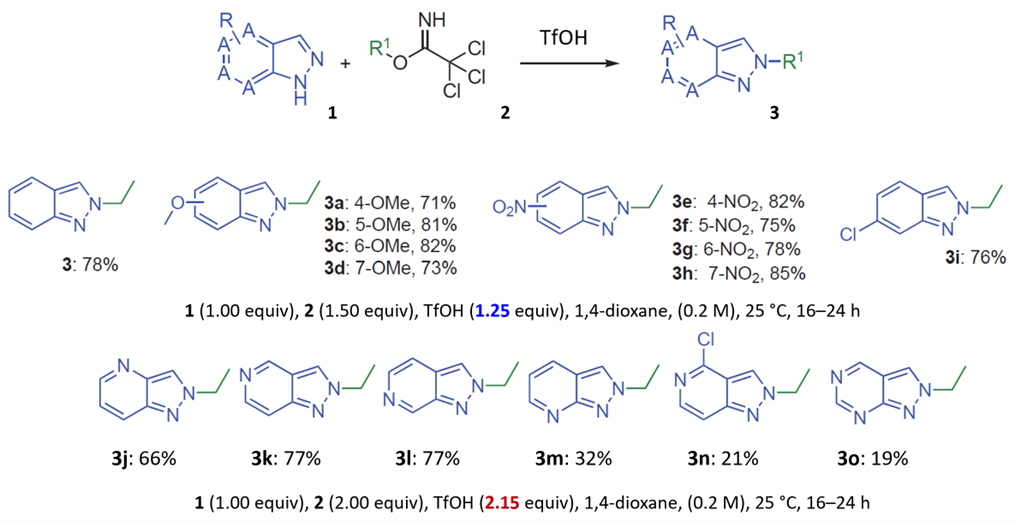
For azaindazole substrates, the article mentioned that limited or no reaction was observed using 1.25 equivalents of TfOH[1]. However, when TfOH was increased to 2.15 equivalents, the N2 alkylation products were selectively formed. The authors proposed that the additional acid preferentially protonates the more basic nitrogen atoms in the pyridine or pyrimidine rings, thereby increasing the Lewis basicity of the N2 nitrogen[1]. Intuitively, protonation would render a nucleophile less reactive. We decided to apply the model developed above to look for more relevant explanations.
We’d like to understand why alkylation of azaindazoles with 1.25 equivalents of TfOH failed. There appear to be two ways to protonate the reacting species:
1. TfOH protonates the imidate and then reacts with the azaindazole. The calculated activation energy for the N2 methylation of azaindazole is 14.89 kcal/mol, only ~1 kcal/mol more than that of indazole. This could not account for the failed alkylation (Figure 11).
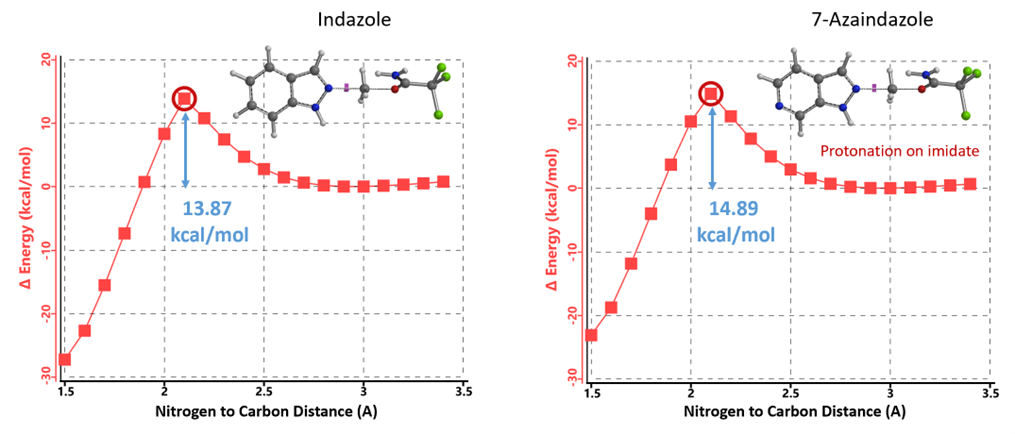
Figure 11. REP of N2 alkylation of protonated imidate with indazole vs 7- azaindazole
2. TfOH protonates the azaindazole and then reacts with the unactivated imidate.
Among the two possible sites for mono-protonation, equilibrium geometry calculation indicates that protonation on the sp2 nitrogen is energetically more favorable than at the imine nitrogen, with a significant energy difference of 9.54 kcal/mol.
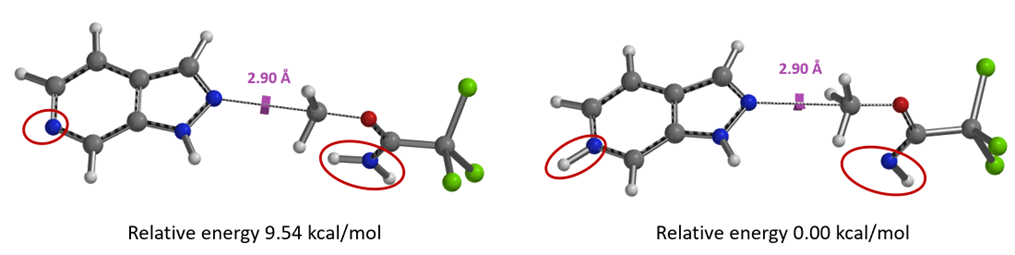
Figure 12. Relative energy of mono-protonation: on the imine N vs the sp2 N
The reaction energy profile for the alkylation of protonated azaindazole with unactivated imidate (Figure 13, right) indicates that the energy barrier of the reaction is 39.98 kcal/mol. This is 26.11 kcal/mol more than that with indazole, accounting for why the alkylation with azaindazole failed. When 1.25 equivalent of TFOH is used, it is the sp2 pyridine nitrogen that gets protonated, leading to a decrease in the reactivity of azaindazole and the imidate is not activated for reaction.
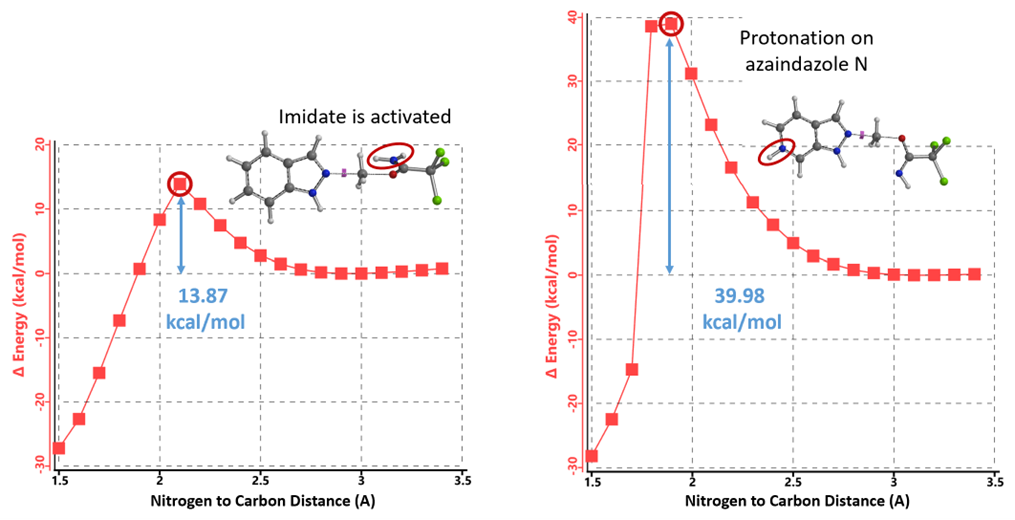
Figure 13. REP of N2 alkylation of protonated imidate with indazole vs unactivated imidate with protonated azaindazole
When the amount of TfOH is increased to 2.15 equivalents, it becomes possible for both the azaindazole and the imide ester to be protonated. The calculated energy barrier of 18.37 kcal/mol (Figure 14, right) suggests that the reaction is achievable. Despite the reduced reactivity of the protonated azaindazole, the activated imidate remains sufficiently reactive for alkylation to occur. The higher energy barrier is consistent with our observation that the reaction proceeds better when elevated from room temperature to 60 °C. Consequently, the original hypothesis posited by the authors—that the additional acid selectively protonates the more basic pyridine/pyrimidine nitrogen, thereby enhancing the Lewis basicity of the N2 nitrogen[1]—does not hold up under these findings.
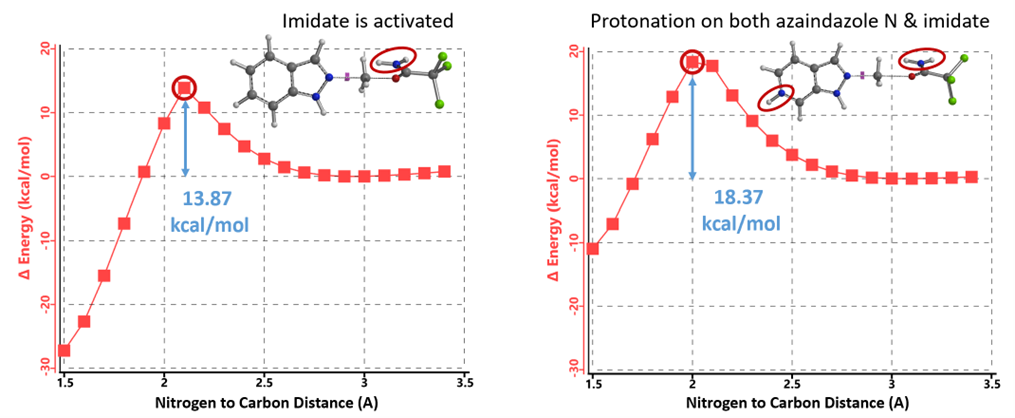
Figure 14. REP of alkylation of protonated azaindazole with protonated activated imidate
Summary
In this chapter, we used QM calculations to investigate why alkylation of indazole with imidate is highly N2 selective and to understand why the size of C3 substituent on the indazole ring has a negligible impact on this selectivity. When evaluating competing reactions involving tautomers, it is essential to include their relative energy difference into consideration. However, it should be noted that estimating the relative energies of tautomers may not be precise enough. In contrast, comparing the energies of their transition states offers a more dependable approach for correlation with experimental observations.
Building on What We Just Learned
For methylation of methyl indazole-7-carboxylate, based on the calculated results shown in Figures 15 and 16, will it be N1 or N2 selective?
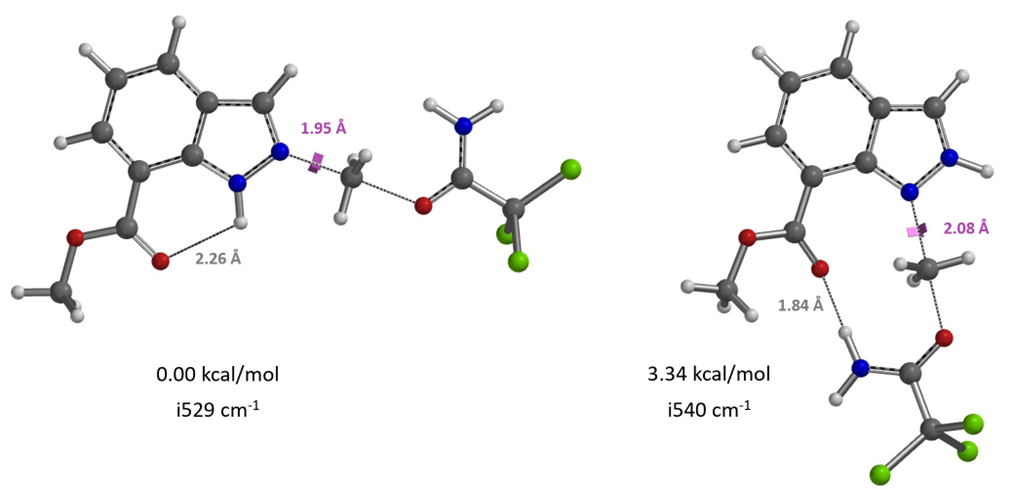
Figure 15. N1 and N2 alkylation transition states of methyl indazole-7-carboxylate
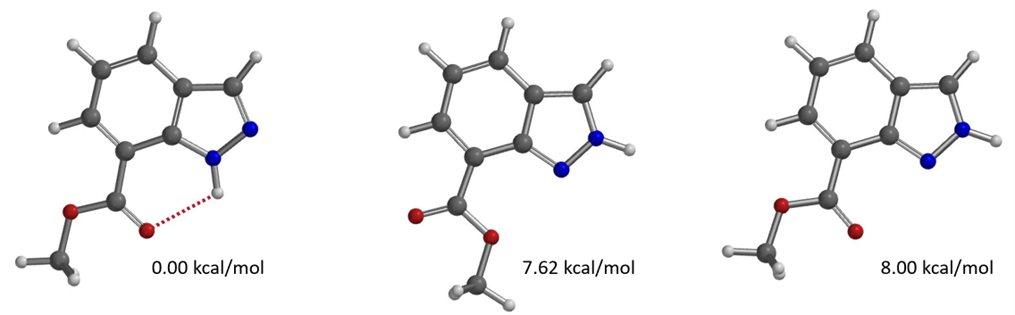
Figure 16. Relative energy of tautomers of methyl indazole-7-carboxylate
[1] J. Clemens, E. L. Bell, A. T. Londregan, Synthesis 2022, 54, 3215.
[2] The Pfizer chemists believe that when R is a primary or secondary alkyl group, the reaction proceeds via an SN2 reaction mechanism. However, when R is a tertiary alkyl group, considering the steric hindrance of the tert-alkyl group, the reaction likely proceeds via a tert-alkyl cation.
[3] Chapter 9 on QM analysis of regioselectivity of alkylation of pyrazole.
[4] a) Spartan’20 Tutorial and User’s Guide (2020). Irvine, CA, USA: Wavefunction, Inc. pp. 158, 442, 459 & 536.
b) Chapters 22 and 24 on the calculation of transition states and imaginary frequencies.
[5] Chapter 49 on Tautomers.
[6] R. Ruzziconi, S. Spizzichino, L. Lunazzi, A. Mazzanti, M. Schlosser, Eur. J. Chem. 2009, 15, 2645 “B values as a sensitive measure of steric effects.”
[6] P. J. Costa, Phys. Sci. Rev. 2017, 2, 136.
[8] Figure 9. Electrostatic potential surfaces are from transition state calculation, with a bond distance of 2.07 Å between indazole N2 and imidate CH3O carbon. The indazole and imide ester were artificially separated further to show the ESP surfaces.
[9] Chapter 42 on visualizing non-covalent interactions with electron density maps.
