
Ketalization with Tetramethoxysilane: QM Mechanism Investigation
QM Magic Class | Chapter 16
< Magical Power of Quantum Mechanics
In organic synthesis, ketalization is a very useful strategy for protection of ketones. This is usually accomplished by treatment of the ketone with a diol in the presence of an acid catalyst. Protection of sterically congested ketones could be challenging, usually requires high reaction temperature, long reaction time, yet still stalls at partial conversion. Even though ketalization with tetramethoxysilane was reported in the literature,[1] experimental conditions are lacking. We found that treatment of these ketones in neat tetramethoxysilane in the presence of p-toluenesulfonic acid could provide almost quantitatively the corresponding dimethoxy ketal (Figure 1).
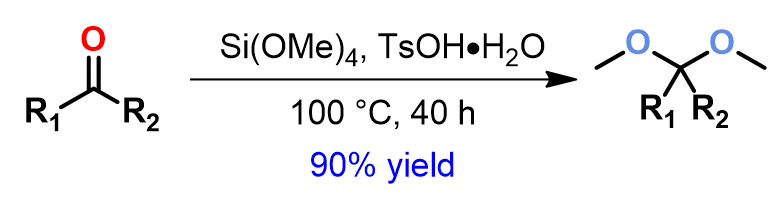
Plausible Mechanism of Reaction
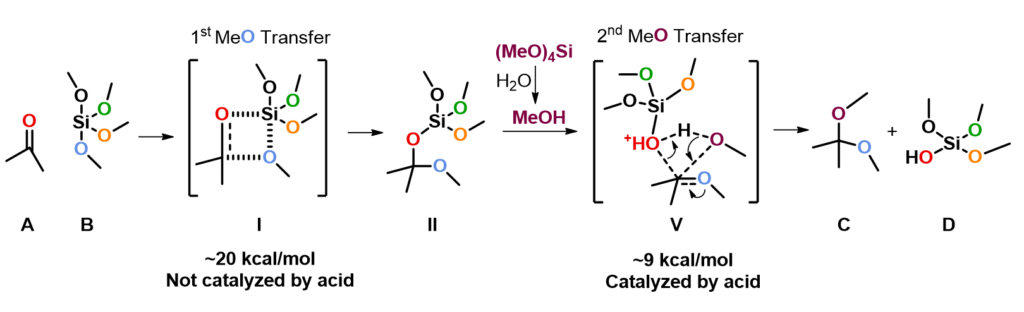
Since mechanism of this reaction has not been reported so far, we decided to study this with QM calculations. We reasoned that the reaction proceeds with the following mechanism (Figure 2). First, tetramethoxysilane transfers a methoxy group to the carbonyl carbon of ketone via a four-membered ring transition state I, resembling mechanism of a Wittig reaction, to give intermediate II, which is then protonated to provide III. Cleavage of the C-O bond of III generates oxonium intermediate IV and (MeO)3SiOH. Then a second methoxy group is transferred from the silyl reagent to IV to provide the ketal product C and dimethyl silicate D.
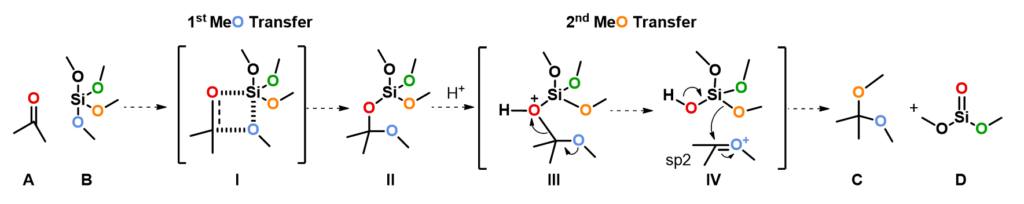
Modeling of the First Methoxy Group Transfer
We modeled reaction energy profile of the first methoxy group transfer from tetramethoxysilane to ketone, with Si approaching carbonyl O from 3.6 Å to 1.7 Å and silyl O approaching carbonyl C from 3.3 Å to 1.4 Å, concurrently with a step size of 0.1 Å, via a four-membered ring transition state I as shown in Figure 3 [2]. The energy maximum of the reaction energy profile is approximated as the transition state of the reaction, translating to an activation energy of 20.7 kcal/mol [3, 4].
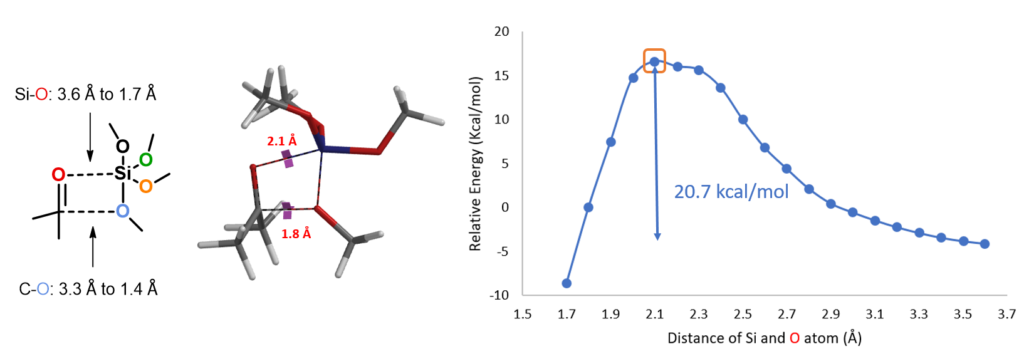
Modeling of the Second Methoxy Group Transfer
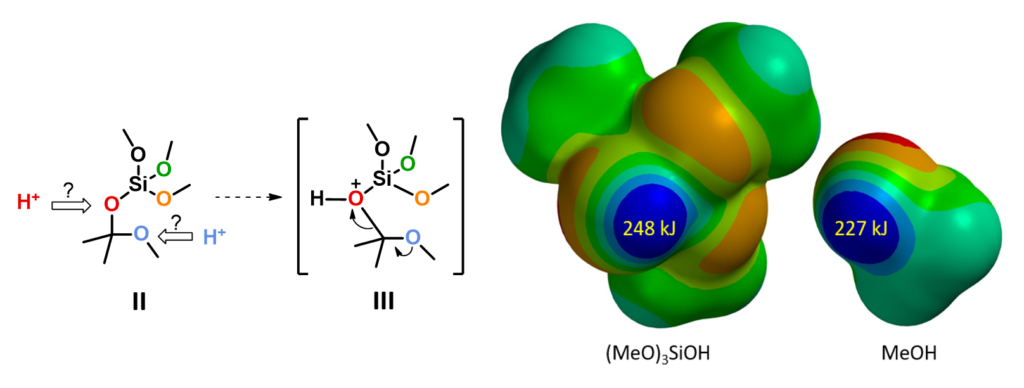
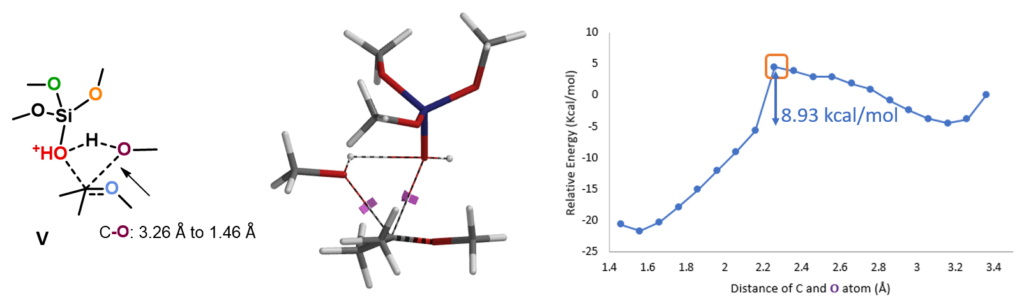
For the next step, electrostatic potential map calculations showed that (MeO)3SiOH is more acidic than methanol (Figure 4, right), and as such (MeO)3SiO is a better leaving group. This suggests that the reaction will proceed through C-O cleavage of the O-protonated intermediate III (Figure 4, left).[5]
The following step will be the transfer of the second methoxy group (Figure 5). But where does the methanol come from? Could it be from decomposition of (MeO)3SiOH?
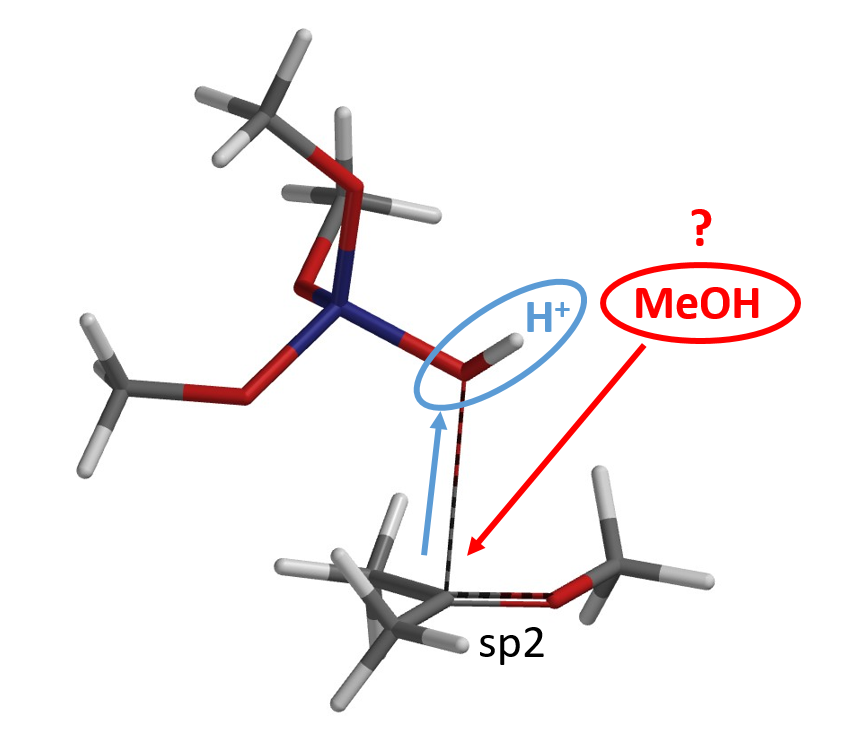
Reaction energy profile calculation showed that decomposition of (MeO)3SiOH to dimethyl silicate and methanol could have a very high energy barrier of 64 kcal/mol that is not likely to be happening under our reaction conditions (Figure 6).
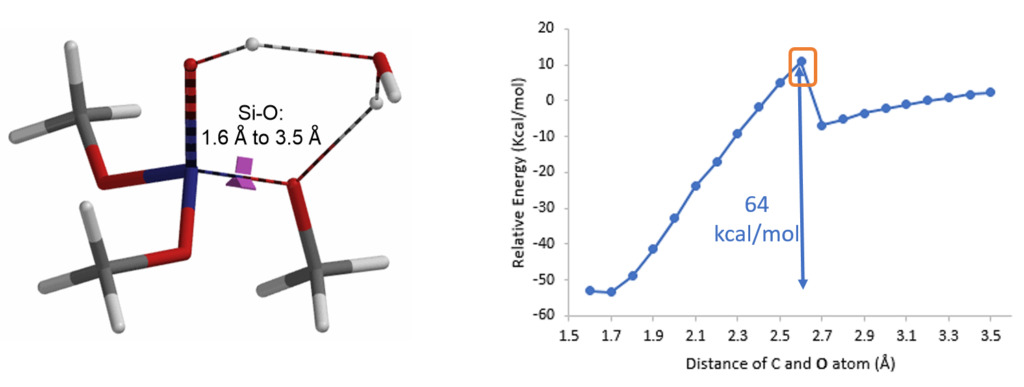
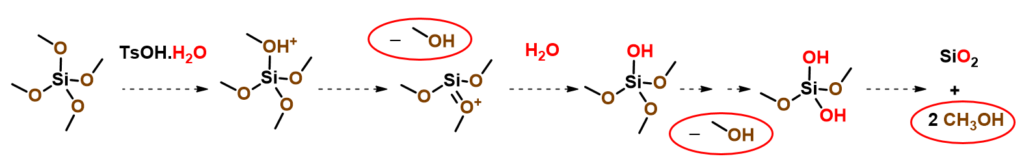
So where could the methanol come from? It becomes obvious that methanol could come from reaction of tetramethoxysilane with water in the hygroscopic p-toluene-sulfonic acid added to the reaction mixture (Figure 7).
With the source of second MeO group settled, we are ready to model the next methoxy transfer step. We reasoned that a concerted mechanism, in which cleavage of the C-O and formation of the C-O bonds proceeds concurrently, are more likely. Indeed, modeling of this reaction shows a relatively low activation energy of 8.93 kcal/mol[3] (Figure 8), a very facile transfer process.
Based on the above calculation results, we updated our proposed mechanism as shown on Figure 9. Ketone A and tetramethoxysilane B react, through a four-membered ring transition state I, to give intermediate II, with an activation energy of ∼20 kcal/mol. Reaction of protonated II and methanol via transition state V with a concerted mechanism producing ketal C, with a relatively low activation energy of ∼9 kcal/mol. Hygroscopic p-toluenesulfonic acid monohydrate catalyzes the second methoxy transfer step and is also the source of methanol, through hydrolysis of tetramethoxysilane.
Experimental Procedure
Reaction conditions for ketalization with tetramethoxysilane is not available from the literature, below are conditions we identified:
A mixture of ketone A (1.0 mmol, 1.0 eq.), tetramethoxysilane (2.5 mL), and TsOH • H2O (0.04 mmol, 0.4 eq.) was stirred under an atmosphere of nitrogen for 40 h at 100 °C. The product mixture was diluted with ethyl acetate and washed with brine (3 times). The organic layer was dried and concentrated under vacuum. The residue was purified by column chromatography on silica gel to provide the ketal product C.
The resultant ketals could be readily hydrolyzed to corresponding ketones under very mild conditions (TFA/CHCl3/H2O, 15/15/1 volume, rt, 1 h).
This article is written and edited by Dong Pan, Zhengquan Zhou, Wenfeng Liu, Guqin Shi, Liting Dong, Tommy Lai, and John S. Wai.
References:
[1] a) Nasarow et al. Zhurnal Obshchei Khimii, 1959, 29, 106. b) Sakurai, H. and Love, J.A., “Silicon (IV) methoxide” in Encyclopedia of Reagents for Organic Synthesis, 2009, John Wiley & Sons.
[2] Spartan ’18 Tutorial and User’s Guide (2019). Irvine, CA, USA: Wavefunction, Inc.
[3] Reactions with activation energies less than 10 kcal/mol can usually proceed at room temperature or lower. For those with activation energies between 10 and 20 kcal/mol, they generally require heating. Reaction with activation energy >20 kcal/mol usually requires higher reaction temperature and longer reaction time.
[4] Attempts to model the first methoxy transfer with ketone carbonyl oxygen protonated were futile.
[5] Since (MeO)3SiO is a better leaving group than MeO, we reasoned that this step proceeds through protonated intermediate III. This is analogous to what we observed with aminal.
