
Advanced Screening Methods for Better Drug Targets

From Undruggable to Actionable: Advanced Screening Methods for Better Drug Targets
Introduction:
Identifying a high-quality drug target is the cornerstone of successful drug discovery, yet it remains one of the most resource-intensive stages of the process. Bringing a new drug to market takes an estimated 5 to 15 years, and selecting the wrong target early on can lead to costly late-stage failures [1,2]. A viable target must meet strict criteria: it must be “druggable,” exhibit a distinct expression pattern in disease states to minimize systemic toxicity, and offer favorable intellectual property prospects. In this article, we discuss cutting-edge drug discovery techniques for target deconvolution, target discovery, and mechanism of action assessment.
The Power of MS-Based Drug Screening
Mass spectrometry (MS) has evolved into a versatile tool for early-stage discovery, including mechanism-of-action (MOA) studies, target deconvolution, and biomarker identification. A standout technique in this field is affinity selection mass spectrometry (ASMS), which offers exceptional throughput for drug screening.
Unlike DNA-encoded libraries (DELs) that require compounds to be physically attached to DNA sequences, ASMS is a label-free method compatible with any drug-like compound library. The ASMS workflow utilizes a size-exclusion chromatography (SEC) column for the initial separation of protein-bound complexes, followed by a reverse-phase (RP) column to release and identify the specific binders via high-resolution mass spectrometry (Figure 1) [1,2].
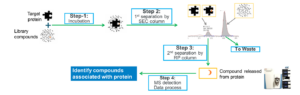
Figure 1. ASMS screening process. [1,2]
Covalent Screening: Overcoming the “Undruggable” Challenge
Covalent drugs, which form irreversible bonds with their targets, have seen a resurgence due to their low-dose requirements and high potency [3]. Historically, researchers avoided covalent molecules due to concerns about off-target toxicity, but modern chemical proteomics now allows researchers to evaluate and mitigate these risks early in the R&D cycle.
For example, KRAS was long considered an “undruggable” target, with the G12C mutation being highly prevalent in many cancers [4]. However, in-cell covalent screening of KRAS-G12C inhibitors demonstrated a near 100% binding ratio. Across multi-omics profiling, the inhibitors consistently showed a clear dose-response curve with an IC50 of approximately 10 nanomolar, confirming high potency and high target selectivity (Figure 2).
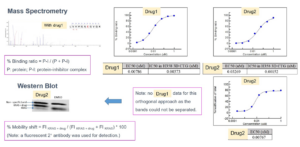
Figure 2. In-cell covalent drug screening. Mass spectrometry confirms a nearly 100% binding ratio. Dose-response curves show highly potent inhibitors with EC50 ~10nM.
Decoding Mechanism of Action and Resistance
Beyond initial screening, proteomics provides a system-wide view of how a drug impacts cellular pathways. While many drugs do not change the total abundance of a protein, they often alter post-translational modifications (PTMs), such as phosphorylation, glycosylation, and acetylation.
- Phosphoproteomics: For KRAS inhibitors, phosphoproteomics revealed the downregulation of ERK phosphorylation, confirming the drug’s efficacy and mapping the specific signaling pathways affected [4].
- Resistance mechanisms: Proteomics is critical in understanding why patients develop resistance to therapies like antibody-drug conjugates (ADCs). In one instance, proteomics profiling showed a 30-fold decrease in target antigen expression and a 1,000-fold increase in a protein that likely hindered ADC internalization [5].
Phenotypic Screening for Novel Target Identification
When a ligand for a disease target is unavailable, target discovery via functional screening becomes essential. WuXi Biology utilizes both in vitro and in vivo screening models to identify key factors that modulate disease phenotypes.
In a study, researchers used gradient host immunity pressure models to
identified key modulators of cancer immunity [6]. By comparing immunodeficient (nude) and immunocompetent (wild-type) mice, they identified ANKRD52 as a key modulator. Interestingly, while ANKRD52-null cells grew poorly in immunodeficient mice due to intrinsic defects, this growth disadvantage was reversed in immunocompetent (wild-type) mice. This indicates that the loss of ANKRD52 confers a survival advantage by helping tumors evade immune surveillance, effectively offsetting their intrinsic growth deficiency (Figure 3).

Figure 3. The result for gradient host immunity pressure phenotypic screening & validation of in vivo model
Target Deconvolution: Proximity Labeling Strategies
Phenotypic screening is often used for first-in-class discovery, but the challenge is determining the actual protein target [7,8]. Proximity labeling involves constructing cell lines with fusion proteins (target + biotin ligase) to label binding partners, such as E3 ligases, that come into close proximity. Using the reference compound ARV110, this technology successfully identified Cereblon (CRBN) as the top-ranked E3 ubiquitin ligase interactor [9].
Future Directions: Cell Painting and MOA Prediction
One of the most innovative technologies in development is cell painting, a high-content image-based approach that records morphological changes in cells using multiplexed fluorescent dyes. By analyzing features such as nuclear size or mitochondrial distribution, researchers can predict a drug’s MOA with high reproducibility (Figure 4A).
In internal validation studies using phenotypically positive compounds from an approved metabolism drug library, 17 out of 22 compounds (~77% accuracy) displayed clustering patterns that aligned perfectly with their known biological pathways. This demonstrates that high-content morphological fingerprints from phenotypic profiling have the potential to reliably predict a drug’s MOA in a high-throughput, phenotypic format (Figure 4B) [10].


Figure 4. Cell painting and UMAP clustering. A. Multiplexed fluorescent staining targets different cellular features. B. UMAP analysis shows that 77% of compounds cluster correctly by pathway.
Conclusion
The path from target discovery to therapeutic validation is complex. To navigate these complexities, WuXi Biology has developed an integrated platform that combines high-throughput screening, advanced chemical proteomics, and functional analysis to accelerate target identification and validation, featuring
- 1,800+ animal model datasets
- 100+ targets successfully discovered and validated for clients over 15 years
- 50+ ready-to-use stable cell lines
By providing end-to-end support—from sequence design and synthesis to targeted delivery and functional validation—we empower our partners to bring revolutionary medicines to patients faster and more reliably.
References
- Whitehurst, C.E. & Annis, D.A. (2008). ASMS Screening Overview. Combinatorial Chemistry & High Throughput Screening, 11, 427-435.
- O’Connell, T.N., et al. (2014). ASMS in Analytical Chemistry. Analytical Chemistry, 86, 7413-7420.
- RSC Medicinal Chemistry. (2020). A Historic Overview of Covalent Inhibitors. RSC Med. Chem., 11, 876–884.
- Huang, et al. (2021). KRAS Mutations in Cancers. Signal Transduct. Target Ther., 6.
- Science Reports. (2024). Global Proteomics vs. Transcriptomics in Resistance Profiling.
- Song, T.Y., et al. (2021). Tumor evolution selectively inactivates the core microRNA machinery for immune evasion. Nature Communications.
- Advanced Science. (2023). Target Deconvolution in Phenotype-based drug discovery. Adv. Sci.
- Drug Discovery Today. (2020). Target identification methods. DOI: 10.1016/j.drudis.2020.09.016.
- Journal of Medicinal Chemistry. (2024). Proximity Labeling Case Study (Cereblon/ARV110). J. Med. Chem.
- Acta Pharmaceutica Sinica B. (2025). Future Directions in Target Deconvolution. 15, 737e756.
Related Content
Cognitive dysfunction affects over 50 million individuals worldwide, with Alzheimer’s disease (AD) representing two-thirds of cases. Due to the complexity...
VIEW RESOURCETarget deconvolution, particularly when paired with proteomics techniques, plays a crucial role in drug discovery and target identification. By using...
VIEW RESOURCE