

Empowering Organic Syntheses with Quantum Mechanics Analyses

Syntheses of specifically functionalized molecules continue to be rate limiting steps (RLS) in drug discovery. Improvement in this RLS means investing only in reactions/synthetic sequences that are more likely to succeed. Structural diversifications, different substitution patterns or unique heterocyclic systems can often adversely impact the crucial step(s) of established synthetic sequences or development of feasible routes. Furthermore, unique arrays of functionalities in novel heterocyclic systems can lead to unexpected reactivities.
Our retrospective analyses of interesting experimental observations with quantum mechanics (QM) calculation provided tremendous insight why certain substrates behave in unique manners. These analyses have changed our perspectives on applications of QM in syntheses.
Foundation of QM is Schrödinger’s wave equations established in the 1920s. Subsequent development of the Frontier Orbital and Conservation of Molecular Symmetry theories in the 1950s and 1960s, respectively, enabled analyses of organic reactions with QM.
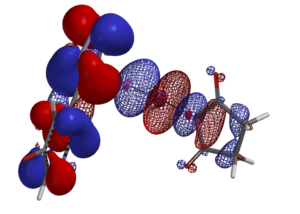
With powerful workstations, state of the art software, and our expertise on reaction QM analyses, real-time incorporations of such calculations in prospective analyses become routines in WuXi AppTec . Our research teams had been invited to present these analyses at the American Chemical Society National Meeting, laboratories of global top universities and pharmaceutical companies.
Our teams are applying and developing QM calculations in the following areas:
Predicting feasibility of reactions
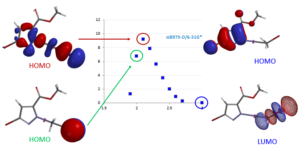
For reactions which exhibit excellent correlations on how well they proceed versus calculated energy gaps between HOMO and LUMO, threshold values could be readily defined. These values are invaluable in assigning feasibility scores for critical reactions in synthetic sequences, and then probability of success (POS) for the whole sequences. Our retrospective and prospective analyses help us to adjust substrates for the reactions, avoid predictable dead ends, reduce cycle times, and improve operation efficiencies.
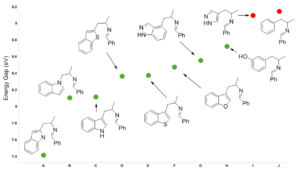
Predicting feasibility and regio-selectivity of nucleophilic reactions
For the nucleophilic substitution reactions, LUMO, LUMO map and Electrostatic Potential map, etc. of the substrates are calculated. When there are potentially multiple reaction sites in the substrates, such information is extremely valuable to predict with high accuracy the regio-selectivity of nucleophilic substitution reactions.
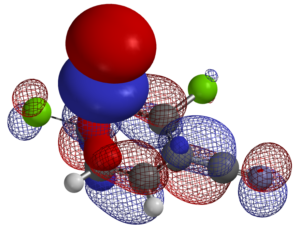
Predicting regio-selectivity of electrophilic reaction of hetero-aromatic systems
For the electrophilic substitution reactions, such as halogenation, we calculate HOMO, NMR, etc. of the reacting substrates. This is an extremely powerful tool in the design of synthetic sequences involving serial halogenation and metal catalyzed coupling reactions.
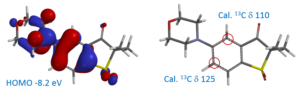
Predicting selectivity of oxidative-addition with poly-halogenated hetero-aromatic substrates
For poly-halogenated substrates, comparisons of calculated IR stretching frequencies and LUMOs have been very informative on which C-X bond is likely to undergo oxidative addition first, and whether the reaction could be selective.
Predicting relative rate of competing paths of reactions
For reactions with competing paths, comparison of the calculated activation energies allow us to predict whether the desired isomers are intrinsically the major products. or to generate insights on how to adjust reaction conditions or sequences.
Our teams continue to expand the scope, depth, and predictive power of QM for organic chemistry. Learn More.
If you are interested or have a need for such study, you can contact us for more information.
Related Content
Delivering comprehensive end-to-end solutions for creating, identifying, and supporting preclinical candidates, from discovery to development, with our integrated chemistry and...
VIEW RESOURCE